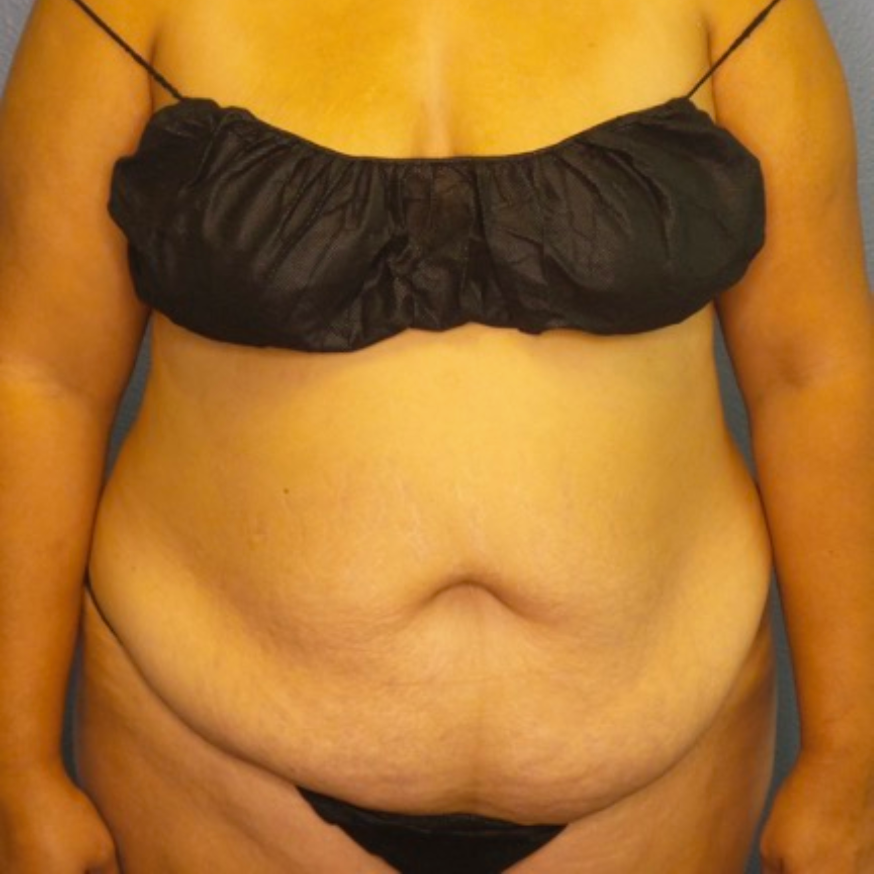
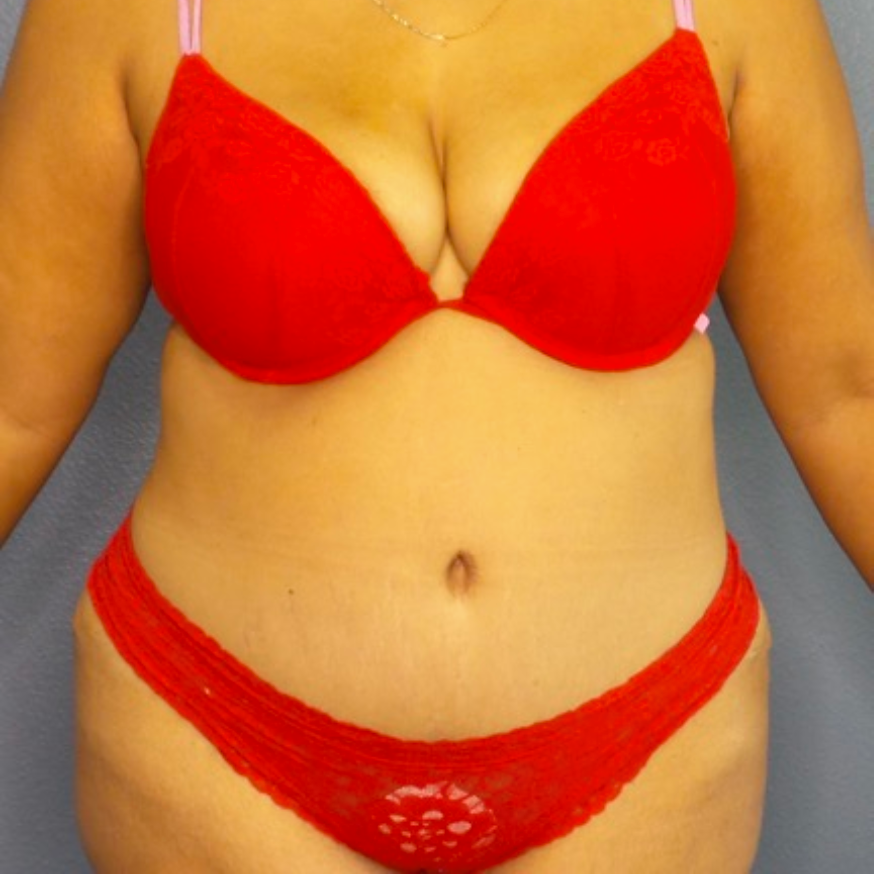
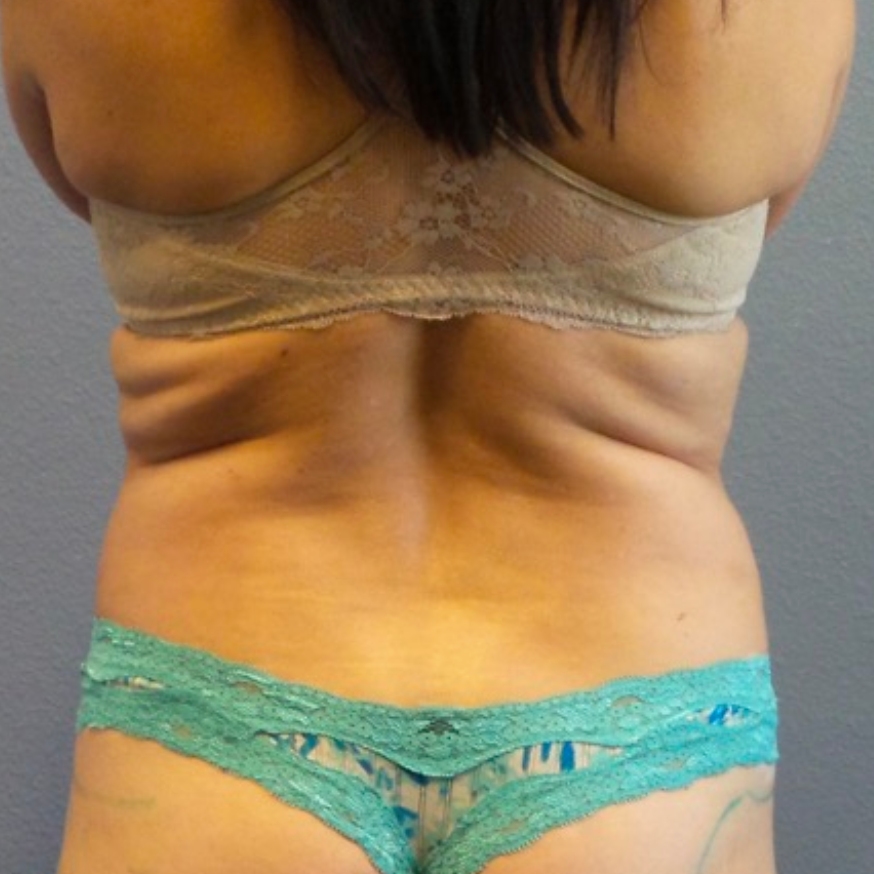

Breast Augmentation Repair
Conveniently located to serve the areas of Tampa and Lakeland, FL
Contents
- 1 FAQ
- 1.1 Will I be able to breast feed after breast enlargement surgery?
- 1.2 Is delayed wound healing common after breast enlargement surgery with silicone breast implants?
- 1.3 What is the Keller Funnel, and what are the advantages of using the device during breast augmentation repair?
- 1.4 What is a periareolar incision in breast enlargement repair surgery?
- 1.5 What is revision breast enlargement or revision breast augmentation surgery?
- 1.6 What type of implants should I ask my surgeon to use in repairing my breasts through revision augmentation surgery?
- 1.7 What is palpable or visible breast implant rippling after revision breast augmentation surgery, what are the consequences, and what is the treatment?
- 1.8 How can the palpability of a breast implant be reduced in breast enlargement repairative surgery?
- 1.9 How much does revision breast enlargement surgery hurt?
- 1.10 Is abscess formation common after revision breast augmentation surgery, what are the consequences, and what is the treatment?
- 1.11 Is infection common after breast enlargement revision surgery with silicone breast implants, what are the consequences, and what is the treatment?
- 1.12 Should I have breast augmentation revision using silicone or saline prosthesis?
- 1.13 Smooth vs. Textured
- 1.14 Subglandular versus Submuscular versus “Dual-Plane” Breast Implant Placement
- 1.15 What type of anesthesia is typically used to carry out revision breast augmentation or repair breast enlargement surgery?
- 1.16 Is it better to use a low profile breast implant or a high profile breast prosthesis in breast enlargement repair-revision surgery?
- 1.17 Should I have breast augmentation revision using textured or smooth breast implants?
- 1.18 Is breast tissue or skin flap death (necrosis) common after revision breast augmentation surgery?
- 1.19 Am I a good candidate for the use of silicone gel-filled breast implants in my revision breast augmentation surgery?
- 1.20 Is breast tissue or skin flap death (necrosis) common after revision breast augmentation surgery with silicone breast implants?
- 1.21 Is seroma (fluid accumulation as your body attempts to fill a potential space) common after revision breast enlargement surgery with silicone breast implants, what are the consequences, and what is th
- 1.22 Is pain normal after revision breast augmentation surgery with silicone implants?
- 1.23 Is loss of nipple sensation possible after breast augmentation revision surgery?
- 1.24 What is implant exposure or breast implant extrusion after revision breast enlargement surgery with silicone breast implants, what are the consequences, and what is the treatment?
- 1.25 Is delayed wound healing common after revision breast enlargement surgery with silicone breast implants?
- 1.26 What can I expect in the post-operative period following revision breast enlargement surgery?
- 1.27 Is abscess formation common after revision breast augmentation surgery with silicone breast implants, what are the consequences, and what is the treatment?
- 1.28 Is breast augmentation revision surgery painful?
- 1.29 Is loss of nipple sensation possible after revision breast augmentation surgery?
- 1.30 Is loss of sensation from the skin of the lower pole of the breast common after revision breast enlargement surgery, what are the consequences, and what is the treatment?
- 1.31 What is an inframammary incision with respect to breast augmentation revision?
- 1.32 What type of incision should I chose when undergoing breast enlargement repair surgery? What are the advantages and disadvantages of each?
- 1.33 How should I care for my incisions after revision breast enlargement surgery?
- 1.34 What is implant exposure or breast implant extrusion after breast enlargement revision surgery, what are the consequences, and what is the treatment?
- 1.35 Is abscess formation common after breast augmentation surgery, what are the consequences, and what is the treatment?
- 1.36 Shaped vs. Round
- 1.37 What is wound dehiscence after breast augmentation repair or revision surgery, what are the consequences, and what is the treatment?
- 1.38 Should my physician use a shaped or round implant in performing my breast augmentation repair?
- 1.39 What is an axillary incision in repair breast enlargement surgery?
- 1.40 Is bleeding common after breast augmentation revision surgery, what are the consequences, and what is the treatment?
- 1.41 Will there be breast skin and breast tissue changes after silicone gel-filled breast implant use in breast augmentation revision surgery?
- 1.42 What other points should I consider when chosing silicone gel-filled breast implants to be used in my breast enlargement revision surgery?
- 1.43 Who would be considered a good candidate for revision or repair of breast enlargement or breast augmentation surgery?
- 1.44 What are the polyurethane breakdown products?
- 1.45 Is the need for additional breast procedures or revision common after breast enlargement surgery?
- 1.46 Minimizing Palpability
- 1.47 What are the various saline implants available from Mentor?
- 1.48 What are Cohesive Gel Breast Implants (Gummy Bear Breast Implants)?
- 1.49 Does the FDA publish any information on breast implants?
- 1.50 What is the customary cost of breast augmentation repair or breast enlargement revision surgery?
- 1.51 Can any surgeon use the cohesive “gummy bear” implant?
- 1.52 Are “gummy bear” implants the final word in breast augmentation?
- 1.53 Will I be able to breast feed after breast enlargement surgery?
FAQ
Will I be able to breast feed after breast enlargement surgery?
Breast augmentation surgery should not affect breast feeding. Because breasts gain more projection and substance after enlargement, they are typically easier to hold. Because they protrude more, and are easier to hold, breast feeding becomes much easier. Paradoxically breast feeding is actually less difficult for some women, after breast augmentation surgery. Given all of this, however, there are studies showing that women with breast implants report an inability to feed in up to 2/3’s of the implanted population, compared to 7 in 100 for women without breast implants. It is doubtful, however, that matching for size and age such results would ever hold up.
Is delayed wound healing common after breast enlargement surgery with silicone breast implants?
Delayed wound healing is not very common after breast enlargement surgery. Because the incisions are relatively small, and with proper technique the breast tissue is not devascularized the problem is rarely seen in healthy patients. Nicotine use, poorly controlled diabetes, chemotherapy, radiation therapy, vascular disease, immunosuppressive therapy or disease, use of corticosteroids may all lead to delayed wound healing.
What is the Keller Funnel, and what are the advantages of using the device during breast augmentation repair?
The Keller Funnel is a cone-shaped device that helps Dr. Gerzenshtein position breast implants. The tip is inserted through the breast implant incision, and the implant is put into the slick funnel. Dr. Gerzenshtein uses a gentle push/squeeze motion to coax the implant into the correct position. One advantage to using the Keller Funnel is its no-touch sterile approach that reduces the risk of infection. Also, the device allows Dr. Gerzenshtein to use minimal amount of force when positioning the implants, putting less stress on the implant shell and decreasing the risk of the implant weakening or rupturing. This can reduce the chance of the implants needing to be removed and replaced again. A surgery using the Keller Funnel is shorter and requires smaller incisions and less tissue trauma than traditional breast augmentation repair.
What is a periareolar incision in breast enlargement repair surgery?
The periareolar incision goes around the nipple. It is less noticeable, smaller than the inframammary incision, and if a lift is required later in time, it may be utilized again. Its disadvantages in breast enlargement surgery are a higher infection rate (because the cut goes through breast tissue), possible difficulty in breastfeeding, and the risk of decreased sensation in breast skin and nipple sensation.
What is revision breast enlargement or revision breast augmentation surgery?
Revision breast augmentation or enlargement surgery is an operation in which the previosuly operated breasts are improved in appearance by correcting asymmetry, unfavorable scarring, or making changes to the shape or size of the previoslu placed breast implants. It really is a breast augmentation repair. The implants may be saline or silicone. The implant is also known as a mammary prosthesis. It typically takes from one half hour to one and a half hours to place the breast device, depending on such things as pocket location, incision, and the type of implant used.
What type of implants should I ask my surgeon to use in repairing my breasts through revision augmentation surgery?
There are multiple different types of implants available for use in breast enlargement. In addition multiple permutations of the various prosthesis types yield a fairly wide array of choices in terms of material, shape, coating, size. In addition, each of the two currently approved breast prosthesis manufacturers in the United States (Inamed™ and Mentor™) makes their own version of each implant with the varying characteristics.
The availabilities are as follows. Each implant is encased in a silicone elastomer shell. The shell may be smooth, or textured. The shell may be shaped like a tear-drop, also referred to as anatomical, or the shell may be perfectly round. Each configuration of the round implant may have a different degree of projection even if the amount of volume contained within is the same. As an example, one style of implant may have a volume of 500cc and a projection of 3.6cm, while another, may have a volume of 500cc but a projection of 4.7cm as a result of a narrower base. The filling material of a breast implant may be silicone, or saline, or both in the case of some implants used for breast reconstruction. The size of an implant may vary to over 1000cc, though volumes of that size are not currently approved in the U.S. and must be procured through companies in Europe or South America.
Please refer to questions related to each specific type of implant for its advantages and disadvantages. The type of implant to be used in breast augmentation revision surgery will correspond to a precise aspect of dissatisfaction with the previous operation, and may vary according to surgeon preference.
What is palpable or visible breast implant rippling after revision breast augmentation surgery, what are the consequences, and what is the treatment?
Breast implants have fold flaws. Because of the consistency of silicone gel, and predetermined, and optimal filling, the flaws may be less pronounced in such breast implants. Saline breast implants may produce scalloping and folding that is made more pronounced by overfilling, or under-filling breast implants. The visibility or palpability of breast implant rippling is accentuated by lack of breast tissue, fat tissue, or muscle tissue between the breast implants, and their overlying skin. The solutions to the problem include replacement with the latest generation cohesive gel implants or augmentation of the soft tissue covering the breast implants with flaps.
How can the palpability of a breast implant be reduced in breast enlargement repairative surgery?
A breast implant is more likely to be noted on manipulation of the breast when they are too big for the breast and soft tissue present, when they are over, rather than under the muscle, and when they are textured. Ensuring a small enough base width, good soft tissue cover with a submuscular or dual-plane placement, and using smooth breast implants will decrease the risk for this.
How much does revision breast enlargement surgery hurt?
The breast enlargement surgery itself should be painless, whether performed under sedation with local anesthetic, or general anesthesia. It is the postoperative period that some may find brings discomfort. Assuming the postoperative course is without complication, pain from breast augmentation surgery peaks the day after surgery, and diminishes over the course of the following three to four days to be tolerable enough without the use of narcotics. Of course, pain tolerance varies significantly from patient to patient. The 72-96 hrs time frame is a “ballpark” figure, and a reflection of personal experience. Several other important factors are crucial to consider in breast enlargement surgery. Pre-incisional administration of local or dilute local (called tumescent) anesthetic greatly diminishes postoperative breast enlargement surgery pain. Postoperative pain and tenderness can be further affected by breast implant placement position. Sub-muscular placement or “dual plane” placement may cause substantially more discomfort than sub-glandular placement because of muscle dissection. If a skin excision is necessary, pain may be more pronounced. The injection of local anesthetic at the conclusion of breast augmentation surgery greatly diminishes postoperative discomfort. In addition, a more effective multiple intercostal nerve block may be performed. Finally, a small catheter may be placed, within the breast implant pocket, for the purpose of delivering local anesthetic in the post breast enlargement surgery period. This has been shown in studies to help significantly with postoperative discomfort. In short, breast enlargement surgery is very well tolerated in most patients.
Is abscess formation common after revision breast augmentation surgery, what are the consequences, and what is the treatment?
Any surgery, in any discipline carries a risk of infection. The risk is calculated based on the degree of contamination for a particular operation. Breast augmentation is considered a “clean” surgery, and carries an overall infection rate of less than two percent. If infection should take place, it will most often affect one or more of three patterns, assuming there is no disseminated spread, and the infection remains localized. Infection can occur in the skin, in the soft tissue surrounding the implant, and in the form of a pus pocket. Skin infection will usually respond to oral antibiotics. Soft tissue infections surrounding the breast implant may respond to oral antibiotics, will sometimes require intravenous antibiotics, and in other cases need to be treated with implant removal. If the infection should progress to, or start out as an abscess (pus-pocket), the only treatment that will be effective in treating the infection and preventing more serious systemic complications is drainage of pus and breast implant removal. Toxic Shock Syndrome (TSS) may result from the presence of a foreign body (breast implant in this case) in the setting of an infection, and is a truly life-threatening condition that needs to be addressed immediately. It is marked by high fever, nausea and vomiting, diarrhea, light-headedness and possibly loss of consciousness, and a diffuse rash. The treatment is timely institution of IV antibiotics, and breast implant removal. If the breast implant is removed, the infection should be treated, the inflammation allowed to resolve, and a new implant placed weeks down the road.
Is infection common after breast enlargement revision surgery with silicone breast implants, what are the consequences, and what is the treatment?
Any surgery, in any discipline carries a risk of infection. The risk is calculated based on the degree of contamination for a particular operation. Breast augmentation is considered a “clean” surgery, and carries an overall infection rate of less than two percent. If infection should take place, it will most often affect one or more of three patterns, assuming there is no disseminated spread, and the infection remains localized. Infection can occur in the skin, in the soft tissue surrounding the implant, and in the form of a pus pocket. Skin infection will usually respond to oral antibiotics. Soft tissue infections surrounding the breast implant may respond to oral antibiotics, will sometimes require intravenous antibiotics, and in other cases need to be treated with implant removal. If the infection should progress to, or start out as an abscess (pus-pocket), the only treatment that will be effective in treating the infection and preventing more serious systemic complications is drainage of pus and breast implant removal. Toxic Shock Syndrome (TSS) may result from the presence of a foreign body (breast implant in this case) in the setting of an infection, and is a truly life-threatening condition that needs to be addressed immediately. It is marked by high fever, nausea and vomiting, diarrhea, light-headedness and possibly loss of consciousness, and a diffuse rash. The treatment is timely institution of IV antibiotics, and breast implant removal. If the breast implant is removed, the infection should be treated, the inflammation allowed to resolve, and a new implant placed weeks down the road.
Should I have breast augmentation revision using silicone or saline prosthesis?
To begin with let’s address the controversy surrounding silicone filled breast implants. There is a well known reporter of Asian descent married to an ex-talk show host who built a career on sensational reporting without any basis in fact, that cheated millions of women out of a perfectly soft, and natural silicone breast augmentation. As is the case with many such issues, the truth was not nearly as well publicized. For a decade, the truth was not made public at all. To add to this, countless cases of alleged harms stemming from the implantation of silicone implants were exploited by immoral attorneys. Since the 1990’s, silicone breast implants have been shown to impart no increase in the incidence of breast, cancer, immune disease, or any other malady so eagerly imparted to them by dishonest litigators, and melodramatic fortune seekers with no regard for the effect it would have on women interested in breast augmentation, and especially augmentation combined with a breast lift. Silicone implants have several drawbacks but in the opinion of many plastic surgeons, such shortcomings are far outweighed by the benefits afforded by their use in breast augmentation. Silicone breast implants are thought to produce a softer breast, less breast contour deformities, and a substantially more natural feel on breast contact than saline breast implants. The drawbacks to silicone mammary prosthesis use are twofold. The first is difficulty in the detection of breast implant rupture. When silicone breast implants rupture, the saline that was used to fill them is reabsorbed, and the discrepancy between what was and what is, or between what the size of the unaffected breast and the side of the affected breast is very obvious. When silicone breast implants rupture, the silicone fill is not absorbed. The change in the affected breast is more consistent with a shape change than a size change. As a consequence, this becomes much more difficult to detect. This would not be a problem, however, silicone incites a significant inflammatory reaction in many patients, leading to a dense capsule, and making it difficult to remove the old breast implant, and achieve a predictable result in placing the new one at the same operation. Staging, or breaking the operation presents the patient with the nuisance of two surgeries. For this reason, a patient with silicone breast implants must be very vigilant in monitoring for signs of implant rupture, as early detection, and re-implantation, makes it much less likely that a significant inflammatory reaction, or a tough breast implant capsule will form.
Smooth vs. Textured
There are advantages and disadvantages to using each type of implant for both the surgeon and the patient. Let’s address the concerns of the patient and then discuss the benefits of one type over another in terms of technical pluses and minuses.
A lot of information on the differences in outcomes between using textured or smooth implants for breast augmentation comes from anecdotal reporting or confounded studies and is thus as useful as preference or hearsay. With that in mind here are the supposed advantages of one over another with respect to different characteristics that affect breast enlargement.
Textured breast implants have a rough surface. The thinking behind this is that if the texture is irregular, cells that form a scar will not as be organized on such a surface, which would result in a softer, more pliable breast capsule, and diminish contracture rates. This disparity in breast capsule formation has never been conclusively demonstrated in studies. Because the textured breast implant does not have a regular surface, it is thicker, but at the same time thought to be weaker, because of its surface flaws. A well-known consequence of using the textured breast implant is its more tenacious adherence to the breast capsule that is laid down around it. This may be an advantage in a woman who has had a teardrop or anatomical implant placed at the time of breast augmentation to hold position. The reason that is important is that the shaped breast implant is not symmetric and must sit in the subglandular or submuscular pocket just so. In a thinner woman, or a woman with minimal native breast tissue, a breast implant that has adhered to the capsule and is not free to move around, will pull on the adjacent breast skin and cause breast surface irregularities. It will also be more palpable, and it may need to be placed through a larger incision owing to its lack of pliability when compared to a smooth breast implant. Textured breast implants may also carry a higher risk of breast implant rupture because any traction or pushing on the breast would also shear the breast implants by virtue of their close association with the breast pocket capsule. Finally, it may be more difficult to remove textured breast implants, again because of their adherence to the breast capsule.
Subglandular versus Submuscular versus “Dual-Plane” Breast Implant Placement
Subglandular placement refers to placement of breast implants under the skin, fat, and breast tissue, but on top of the muscle. As a result of this, patients who are thin, and lack significant breast tissue will have an increased chance for implant palpability. The risk for capsular contracture is also significantly higher for both saline and silicone breast implants when placed over the muscle. The advantage to using this approach is the ability to take up loose skin at the lower poles of the breasts, and avoid the longer incisions necessary for a breast lift in some cases.
Submuscular placement puts the breast implant pocket between the ribs of the chest wall and the chest (pectoralis) muscle on top. It is much less prone to cause breast implant palpability than subglandular implant placement, and is associated with a lower risk of scarring and hardness around the implants. Its disadvantage is the propensity to cause a higher riding breast implant. When the pectoralis muscles are contracted the implants are also prone to move up in a very unnatural fashion.
The “dual-plane” approach allows placement of the upper portion of the implant under the muscle, and by releasing the lower portion of the muscle allows the lower portion of the breast implant to sit under the breast tissue. This eliminates the drawbacks of both the sub-glandular and sub-muscular placement while retaining the advantages of both. It is the most commonly performed placement in today’s breast augmentation surgery.
What type of anesthesia is typically used to carry out revision breast augmentation or repair breast enlargement surgery?
The two options available to clients and surgeons in maintaining a comfortable, safe and painless environment for the breast enlargement patient, and a controlled setting for the breast surgeon performing breast enlargement surgery are general anesthesia and intravenous sedation combined with local anesthesia. The advantages of general anesthesia include complete unawareness on the part of the patient during breast implant placement, a secure airway, and a still, controlled environment for the operating surgeon. The greatest disadvantages are post-operative nausea and vomiting, risks associated with general anesthesia and post-operative lethargy that slow recovery. The advantages to sedation type anesthesia for mammary enlargement surgery are just the reverse of the disadvantages noted for general anesthesia; recovery is faster, nausea and vomiting are minimized, and the systemic risks associated with general anesthesia are abolished. Having stated all of this, it is possible that the degree of repairs to be made to breasts that have been previously enlarged may be too extensive to permit revision without general anesthetic.
Is it better to use a low profile breast implant or a high profile breast prosthesis in breast enlargement repair-revision surgery?
There are several advantages to the use of high profile breast implants. First, just as the name suggests, for any given fill volume, high profile breast implants will implant more projection or profile when compared to moderate or “normal” breast prosthesis, and most certainly more than low profile, or anatomically shaped breast implants. The way to picture this is that if you had a cone with a highly sitting tip versus a cone with a wide base, the narrower, taller cone would point more (think of Madonna’s show bra!) and thus give more projection to the breast. What this means is that women with a narrower, smaller chest wall can still have larger breasts. The advantage to some women, and disadvantage to others comes from a basic difference in perception as what a natural breast should look like. If a patient prefers a highly “perky,” high profile, or well projecting breast, the high profile breast implant would be considered ideal. High profile breast implants would not be ideal to place in a patient who prefers natural, gently sloping, and slightly ptotic (hanging) breasts, or in a client who is large and wide chested. Placing full profile breast implants in the case of a wide chest would impart a very unnatural “double cone” appearance. For women with a mid-range chest-wall diameter the choice is one of partiality. That is, the decision has to be made between projection, and cleavage. This is because lower profile implants with a wide base will naturally fill up the inner, otherwise known as the medial breast, and produce cleavage. Finally, it is mostly the anecdotal opinion of some authorities that full or high profile implants tend to generate less rippling.
Should I have breast augmentation revision using textured or smooth breast implants?
There are advantages and disadvantages to using each type of implant for both the surgeon and the patient. Let’s address the concerns of the patient and then discuss the benefits of one type over another in terms of technical pluses and minuses.
A lot of information on the differences in outcomes between using textured or smooth implants for breast augmentation comes from anecdotal reporting or confounded studies and is thus as useful as preference or hearsay. With that in mind here are the supposed advantages of one over another with respect to different characteristics that affect breast enlargement.
Textured breast implants have a rough surface. The thinking behind this is that if the texture is irregular, cells that form a scar will not as be organized on such a surface, which would result in a softer, more pliable breast capsule, and diminish contracture rates. This disparity in breast capsule formation has never been conclusively demonstrated in studies. Because the textured breast implant does not have a regular surface, it is thicker, but at the same time thought to be weaker, because of its surface flaws. A well-known consequence of using the textured breast implant is its more tenacious adherence to the breast capsule that is laid down around it. This may be an advantage in a woman who has had a teardrop or anatomical implant placed at the time of breast augmentation to hold position. The reason that is important is that the shaped breast implant is not symmetric and must sit in the subglandular or submuscular pocket just so. In a thinner woman, or a woman with minimal native breast tissue, a breast implant that has adhered to the capsule and is not free to move around, will pull on the adjacent breast skin and cause breast surface irregularities. It will also be more palpable, and it may need to be placed through a larger incision owing to its lack of pliability when compared to a smooth breast implant. Textured breast implants may also carry a higher risk of breast implant rupture because any traction or pushing on the breast would also shear the breast implants by virtue of their close association with the breast pocket capsule. Finally, it may be more difficult to remove textured breast implants, again because of their adherence to the breast capsule.
Is breast tissue or skin flap death (necrosis) common after revision breast augmentation surgery?
Necrosis of breast tissue or soft tissues and skin is very unusual after breast augmentation only, it is much more likely in the case of a breast lift where a significant amount of tissue is removed. Necrosis is the result of loss of blood supply, hence oxygen, leading to tissue death. Blood supply is lost when dissection proceeds through tissue containing vessels that supply a given area, and transects them. Since multiple vessels usually supply any given area of breast and skin, sacrifice of a few vessels is usually well tolerated. The use of nicotine in any form (smoking, chewing, gum, patch, etc.), chemotherapy, radiation, corticosteroids, contamination all compromise blood supply and make the sacrifice of even a few vessels a significant risk for breast tissue death.
Am I a good candidate for the use of silicone gel-filled breast implants in my revision breast augmentation surgery?
Silicone implants have several drawbacks but in the opinion of many plastic surgeons, such shortcomings are far outweighed by the benefits afforded by their use in breast augmentation. Silicone breast implants are thought to produce a softer breast, less breast contour deformities, and a substantially more natural feel on breast contact than saline breast implants. The drawbacks to silicone mammary prosthesis use are twofold. The first is difficulty in the detection of breast implant rupture. When silicone breast implants rupture, the saline that was used to fill them is reabsorbed, and the discrepancy between what was and what is, or between what the size of the unaffected breast and the side of the affected breast is very obvious. When silicone breast implants rupture, the silicone fill is not absorbed. The change in the affected breast is more consistent with a shape change than a size change. As a consequence, this becomes much more difficult to detect. This would not be a problem, however, silicone incites a significant inflammatory reaction in many patients, leading to a dense capsule, and making it difficult to remove the old breast implant, and achieve a predictable result in placing the new one at the same operation. Staging, or breaking the operation presents the patient with the nuisance of two surgeries. For this reason, a patient with silicone breast implants must be very vigilant in monitoring for signs of implant rupture, as early detection, and re-implantation, makes it much less likely that a significant inflammatory reaction, or a tough breast implant capsule will form
The ideal candidate for a primary breast augmentation with silicone, therefore, is a patient who is secure with silicone as implantation material, is very aware of the shape of her breasts, and vigilant in monitoring for rupture, has a very small amount of native breast tissue, or has a significant amount of droop and desires sub-glandular placement to correct it with minimal
Is breast tissue or skin flap death (necrosis) common after revision breast augmentation surgery with silicone breast implants?
Necrosis of breast tissue or soft tissues and skin is very unusual after breast augmentation only, it is much more likely in the case of a breast lift where a significant amount of tissue is removed. Necrosis is the result of loss of blood supply, hence oxygen, leading to tissue death. Blood supply is lost when dissection proceeds through tissue containing vessels that supply a given area, and transects them. Since multiple vessels usually supply any given area of breast and skin, sacrifice of a few vessels is usually well tolerated. The use of nicotine in any form (smoking, chewing, gum, patch, etc.), chemotherapy, radiation, corticosteroids, contamination all compromise blood supply and make the sacrifice of even a few vessels a significant risk for breast tissue death.
Is seroma (fluid accumulation as your body attempts to fill a potential space) common after revision breast enlargement surgery with silicone breast implants, what are the consequences, and what is th
In breast augmentation surgery, a space is created under the soft tissues of the chest wall. This is the space where the breast implant is placed. The size of the pocket and the size of the implant are seldom identical, and the discrepancy creates a potential space where fluid can collect. The dissection through the soft tissue to the breast implant pocket also makes a potential space for fluid to collect. Sometimes a long standing blood collection can leave a space after it is resorbed. Most seromae resolve with only a few drainage attempts, without an incision. In cases where post-breast augmentation seromae fail to resolve, lining of the fluid pocket needs to be excised because this is what makes the fluid.
Is pain normal after revision breast augmentation surgery with silicone implants?
The breast enlargement surgery itself should be painless, whether performed under sedation with local anesthetic, or general anesthesia. It is the postoperative period that some may find brings discomfort. Assuming the postoperative course is without complication, pain from breast augmentation surgery peaks the day after surgery, and diminishes over the course of the following three to four days to be tolerable enough without the use of narcotics. Of course, pain tolerance varies significantly from patient to patient. The 72-96 hrs time frame is a “ballpark” figure, and a reflection of personal experience. Several other important factors are crucial to consider in breast enlargement surgery. Pre-incisional administration of local or dilute local (called tumescent) anesthetic greatly diminishes postoperative breast enlargement surgery pain. Postoperative pain and tenderness can be further affected by breast implant placement position. Sub-muscular placement or “dual plane” placement may cause substantially more discomfort than sub-glandular placement because of muscle dissection. If a skin excision is necessary, pain may be more pronounced. The injection of local anesthetic at the conclusion of breast augmentation surgery greatly diminishes postoperative discomfort. In addition, a more effective multiple intercostal nerve block may be performed. Finally, a small catheter may be placed, within the breast implant pocket, for the purpose of delivering local anesthetic in the post breast enlargement surgery period. This has been shown in studies to help significantly with postoperative discomfort. In short, breast enlargement surgery is very well tolerated in most patients.
Having stated all of this, it is important to note that pain should get progressively better. Pain that is persistent, uncontrolled by pain medication, or increases in duration and/or intensity after breast augmentation may be a sign of post-operative complication and requires prompt attention.
Is loss of nipple sensation possible after breast augmentation revision surgery?
Nerves that supply sensation to the nipple and surrounding (areolar) skin may be cut or stretched during surgery. Most often this is not the case, and if sensation is lost, it generally due o stretching of the nerves, and will eventually recover. Permanent loss of nipple sensation, however, is always a possibility. Sometimes the nipple skin will become more sensitive to any form of stimulation, which may be due to nerve involvement, or to the stretching of the skin by the breast implant. This, too, is normally temporary. Usually, it is not the nipple that loses sensation, but the nerves to the skin of the lower part of the breast, especially on its outside edge. This is because the nerves giving sensation to that part are most frequently encountered, and manipulated in some way. As with nipple sensation, this usually returns, but not always.
What is implant exposure or breast implant extrusion after revision breast enlargement surgery with silicone breast implants, what are the consequences, and what is the treatment?
The exact risk percentage may be found for both the Allergan, and the Mentor studies in related questions. The problem most often is the result of inadequate soft tissue (breast and skin) overlying the breast implants. It is unusual in cases of primary augmentation, and is more commonly seen in the compromised soft tissue of the reconstructed breast, with or without a history of radiation. If the extrusion if only threatened, and no implant is actually showing at the time of detection, a salvage procedure may be done to improve the soft tissue cover atop the implant. If the implant is exposed, many plastic surgeons will not attempt salvage, as by definition, and exposed implant is a contaminated implant. The breast implant will be removed, patient allowed to heal, and implant replaced at a later time.
Is delayed wound healing common after revision breast enlargement surgery with silicone breast implants?
Delayed wound healing is not very common after breast enlargement surgery. Because the incisions are relatively small, and with proper technique the breast tissue is not devascularized the problem is rarely seen in healthy patients. Nicotine use, poorly controlled diabetes, chemotherapy, radiation therapy, vascular disease, immunosuppressive therapy or disease, use of corticosteroids may all lead to delayed wound healing.
What can I expect in the post-operative period following revision breast enlargement surgery?
The following information is given to Dr. Gerzenshtein’s patients before surgery to inform them of a typical course after breast augmentation. Your plastic surgeon may have a different working environment, and his or her patients a different experience. Consult with your physician about their impressions of patient experience. On waking from anesthesia, you will find yourself in the recovery room with dressings and/or bra in place. You will be able to depart once sufficiently recovered from anesthesia. A friend or family member should drive you home and stay with you for the next 2 days to help you with activities of daily living. You will feel tired and run down for the first several days after general anesthesia, this will improve substantially over the first week. Discharge should be minimal over the next 48 hours; bleeding may occur with excessive activity. If dilute local solution was used (superwet or tumescent technique) pain and discomfort will be mild initially, it will increase and peak within two days, it will then subside over the course of one to two weeks, please use pain medication as needed to help. Nausea and vomiting in the postoperative period is not uncommon and has to do with the type of anesthesia used, and overall patient sensitivity to the various medications, it generally resolves within one to two days after surgery, increasing fluid intake, especially via one of the “ade” (gatorade, powerade, etc.) solutions available for sports use, combined with anti-emetic medication should minimize this problem. Use of opiate pain medication, combined with inactivity, and dehydration may lead to constipation, increasing fluid intake will help this as well, especially in combination with walking, and the use of the prescribed stool softener. Swelling and bruising peak within three days of surgery and gradually subside over the following week. Healing incisions will adopt a pinkish hue which should gradually fade over the next six months to a year. Some patients react to absorbable (inside) suture, small pustules or whiteheads along the incision may signal this, the suture may be removed in the office if the problems becomes bothersome. Numbness may affect the breast skin, and/or the nipple, most commonly this involves the lower pole of the breast skin, and resolves on its own within six months.
Is abscess formation common after revision breast augmentation surgery with silicone breast implants, what are the consequences, and what is the treatment?
Any surgery, in any discipline carries a risk of infection. The risk is calculated based on the degree of contamination for a particular operation. Breast augmentation is considered a “clean” surgery, and carries an overall infection rate of less than two percent. If infection should take place, it will most often affect one or more of three patterns, assuming there is no disseminated spread, and the infection remains localized. Infection can occur in the skin, in the soft tissue surrounding the implant, and in the form of a pus pocket. Skin infection will usually respond to oral antibiotics. Soft tissue infections surrounding the breast implant may respond to oral antibiotics, will sometimes require intravenous antibiotics, and in other cases need to be treated with implant removal. If the infection should progress to, or start out as an abscess (pus-pocket), the only treatment that will be effective in treating the infection and preventing more serious systemic complications is drainage of pus and breast implant removal. Toxic Shock Syndrome (TSS) may result from the presence of a foreign body (breast implant in this case) in the setting of an infection, and is a truly life-threatening condition that needs to be addressed immediately. It is marked by high fever, nausea and vomiting, diarrhea, light-headedness and possibly loss of consciousness, and a diffuse rash. The treatment is timely institution of IV antibiotics, and breast implant removal. If the breast implant is removed, the infection should be treated, the inflammation allowed to resolve, and a new implant placed weeks down the road.
Is breast augmentation revision surgery painful?
The breast enlargement surgery itself should be painless, whether performed under sedation with local anesthetic, or general anesthesia. It is the postoperative period that some may find brings discomfort. Assuming the postoperative course is without complication, pain from breast augmentation surgery peaks the day after surgery, and diminishes over the course of the following three to four days to be tolerable enough without the use of narcotics. Of course, pain tolerance varies significantly from patient to patient. The 72-96 hrs time frame is a “ballpark” figure, and a reflection of personal experience. Several other important factors are crucial to consider in breast enlargement surgery. Pre-incisional administration of local or dilute local (called tumescent) anesthetic greatly diminishes postoperative breast enlargement surgery pain. Postoperative pain and tenderness can be further affected by breast implant placement position. Sub-muscular placement or “dual plane” placement may cause substantially more discomfort than sub-glandular placement because of muscle dissection. If a skin excision is necessary, pain may be more pronounced. The injection of local anesthetic at the conclusion of breast augmentation surgery greatly diminishes postoperative discomfort. In addition, a more effective multiple intercostal nerve block may be performed. Finally, a small catheter may be placed, within the breast implant pocket, for the purpose of delivering local anesthetic in the post breast enlargement surgery period. This has been shown in studies to help significantly with postoperative discomfort. In short, breast enlargement surgery is very well tolerated in most patients.
Is loss of nipple sensation possible after revision breast augmentation surgery?
Nerves that supply sensation to the nipple and surrounding (areolar) skin may be cut or stretched during surgery. Most often this is not the case, and if sensation is lost, it generally due o stretching of the nerves, and will eventually recover. Permanent loss of nipple sensation, however, is always a possibility. Sometimes the nipple skin will become more sensitive to any form of stimulation, which may be due to nerve involvement, or to the stretching of the skin by the breast implant. This, too, is normally temporary. Usually, it is not the nipple that loses sensation, but the nerves to the skin of the lower part of the breast, especially on its outside edge. This is because the nerves giving sensation to that part are most frequently encountered, and manipulated in some way. As with nipple sensation, this usually returns, but not always.
Is loss of sensation from the skin of the lower pole of the breast common after revision breast enlargement surgery, what are the consequences, and what is the treatment?
Nerves that supply sensation to the nipple and surrounding (areolar) skin may be cut or stretched during surgery. Most often this is not the case, and if sensation is lost, it generally due o stretching of the nerves, and will eventually recover. Permanent loss of nipple sensation, however, is always a possibility. Sometimes the nipple skin will become more sensitive to any form of stimulation, which may be due to nerve involvement, or to the stretching of the skin by the breast implant. This, too, is normally temporary. Usually, it is not the nipple that loses sensation, but the nerves to the skin of the lower part of the breast, especially on its outside edge. This is because the nerves giving sensation to that part are most frequently encountered, and manipulated in some way. As with nipple sensation, this usually returns, but not always.
What is an inframammary incision with respect to breast augmentation revision?
The inframammary approach to revision breast augmentation surgery allows your plastic surgeon the greatest visibility while dissecting an implant pocket, and as such, allows greater precision and control of symmetry. The inframammary incision is placed in the fold under the breast, in the breast fold, and tends to be more noticeable for two reasons. It is typically larger than other types of breast incisions. It has a tendency to migrate as the implant settles up or down, and its final resting place is less predictable.
What type of incision should I chose when undergoing breast enlargement repair surgery? What are the advantages and disadvantages of each?
The inframammary approach to breast augmentation surgery allows your plastic surgeon the greatest visibility while dissecting an implant pocket, and as such, allows greater precision and control of symmetry. The inframammary incision is placed in the fold under the breast, in the breast fold, and tends to be more noticeable for two reasons. It is typically larger than other types of breast incisions. It has a tendency to migrate as the implant settles up or down, and its final resting place is less predictable.
The periareolar incision goes around the nipple. It is less noticeable, smaller than the inframammary incision, and if a lift is required later in time, it may be utilized again. Its disadvantages in breast enlargement surgery are a higher infection rate (because the cut goes through breast tissue), possible difficulty in breastfeeding, and the risk of decreased sensation in breast skin and nipple sensation.
Placing the incision in the axilla (underarm) allows for a smaller incision. It is useful when there is not much breast tissue present to begin with, and thus no prominent breast fold, and when the nipple-areola complex is small. It is not as well concealed as the periareolar incision, gives less control in terms of the ability to feel around the breast pockets in assuring symmetry, and placement of silicone breast implants will necessitate a larger incision which may be less pleasing.
The peri-umbilical incision is placed at the top of the belly-button. Its obvious advantage is a lack of scars on, around, or near the breasts. Its disadvantages are blind dissection, making asymmetry more common, and making the likelihood of one side being submuscular and the other subglandular more likely. If an undesirable result is obtained, a new incision will be needed to correct the problem. It may also damage the breast implants, and cannot be used with pre-filled silicone breast implants.
Unfortunately, in operating on a previously augmented breast, the plastic surgeon can only go through the previous incision, whatever that may have been, or have to place an additional incision with the consent of the patient.
How should I care for my incisions after revision breast enlargement surgery?
Dr. Gerzenshtein’s recommendations to his patients are found below. Consult with your own surgeon for his ore her specific instructions. After surgery you will be placed in a bra, possibly with an ace wrap above it. If the ace is present, it was applied to control pocket position in cases where implant position was made submuscular and the muscle was not entirely divided at its lower edge, this means that the ace wrap must be re-applied exactly the way you were instructed. The ace wrap along with the dressing, will removed for the first time 24 hours after surgery, in the office. After the initial dressing change, dressings should be changed twice a day in the following manner: Remove old dressing and wash or shower, after drying, apply new dressing; ABDs followed by bra, then ace if indicated. When the edges of the steri-strips become frayed trim them. With time, as very little is left behind, they may be removed (usually 2-4 weeks). It is not routine to have drains placed at the time of surgery, however, at times, if bleeding is diffuse, and cannot be addressed via surgical maneuvers (clipping, suturing, tying) it may be safer to leave behind a drain in attempting to prevent a hematoma (blood collection), if present, the drains will be removed within one to three days. Wear a soft comfortable bra for the first 7 days after surgery. Two weeks after surgery you may purchase any type of bra you wish. If non-absorbable sutures were used, they will be removed 7 days after surgery. You may begin breast massage the 2nd week after surgery, you will be instructed on breast massage at your post-op appointment, continue massaging for at least 6 weeks after surgery. Do not expose incisions to the sun and/or tanning UV light for at least 1 year, however, you may begin tanning 4 weeks after surgery while keeping incisions covered. If sun exposure in unavoidable, use a product with SPF of at least 30.
What is implant exposure or breast implant extrusion after breast enlargement revision surgery, what are the consequences, and what is the treatment?
The exact risk percentage may be found for both the Allergan, and the Mentor studies in related questions. The problem most often is the result of inadequate soft tissue (breast and skin) overlying the breast implants. It is unusual in cases of primary augmentation, and is more commonly seen in the compromised soft tissue of the reconstructed breast. If the extrusion is only threatened, and no implant is actually showing at the time of detection, a salvage procedure may be done to improve the soft tissue cover atop the implant. If the implant is exposed, many plastic surgeons will not attempt salvage, as by definition, and exposed implant is a contaminated implant. The breast implant will be removed, patient allowed to heal, and implant replaced at a later time.
Is abscess formation common after breast augmentation surgery, what are the consequences, and what is the treatment?
Any surgery, in any discipline carries a risk of infection. The risk is calculated based on the degree of contamination for a particular operation. Breast augmentation is considered a “clean” surgery, and carries an overall infection rate of less than two percent. If infection should take place, it will most often affect one or more of three patterns, assuming there is no disseminated spread, and the infection remains localized. Infection can occur in the skin, in the soft tissue surrounding the implant, and in the form of a pus pocket. Skin infection will usually respond to oral antibiotics. Soft tissue infections surrounding the breast implant may respond to oral antibiotics, will sometimes require intravenous antibiotics, and in other cases need to be treated with implant removal. If the infection should progress to, or start out as an abscess (pus-pocket), the only treatment that will be effective in treating the infection and preventing more serious systemic complications is drainage of pus and breast implant removal. Toxic Shock Syndrome (TSS) may result from the presence of a foreign body (breast implant in this case) in the setting of an infection, and is a truly life-threatening condition that needs to be addressed immediately. It is marked by high fever, nausea and vomiting, diarrhea, light-headedness and possibly loss of consciousness, and a diffuse rash. The treatment is timely institution of IV antibiotics, and breast implant removal. If the breast implant is removed, the infection should be treated, the inflammation allowed to resolve, and a new implant placed weeks down the road.
Shaped vs. Round
This decision depends as much on what the surgeon can accomplish using either the round or the anatomically contoured implants as on which type of implants to use. Assuming the breast pockets are dissected in to the inferior or lowermost extent in the exact same manner, the shaped implants will impart more fullness at the top, and provide a more natural, gently curving slope to the augmented breast. For this to happen, however, the lowermost portion of the dissection or pocket must be the same no matter which mammary implants you use. If a surgeon habitually dissects low enough inferiorly to drop round breast prosthesis low enough that upper pole fullness is lost, that surgeon will likely favor the teardrop shaped, or anatomically shaped implant to compensate for that dissection. For anatomically shaped or contoured implants to do what they were intended for, pocket dissection has to extremely precise. This is because contoured breast implants are not symmetric; they have a top and bottom. If the subglandular or submuscular pockets are too wide, the shaped implants can shift or even flip, imparting asymmetry and even worse an unnatural shape to the augmented breast. Even though shaped mammary implants are textured, this is still no guarantee against malposition. Finally using the round type of breast implants can impart more medial or inner breast fullness. This significantly improves cleavage, because more fill volume winds up toward the more central part of the breast.
There are several advantages to the use of high profile breast implants. First, just as the name suggests, for any given fill volume, high profile breast implants will implant more projection or profile when compared to moderate or “normal” breast prosthesis, and most certainly more than low profile, or anatomically shaped breast implants. The way to picture this is that if you had a cone with a highly sitting tip versus a cone with a wide base, the narrower, taller cone would point more (think of Madonna’s show bra!) and thus give more projection to the breast. What this means is that women with a narrower, smaller chest wall can still have larger breasts. The advantage to some women, and disadvantage to others comes from a basic difference in perception as what a natural breast should look like. If a patient prefers a highly “perky,” high profile, or well projecting breast, the high profile breast implant would be considered ideal. High profile breast implants would not be ideal to place in a patient who prefers natural, gently sloping, and slightly ptotic (hanging) breasts, or in a client who is large and wide chested. Placing full profile breast implants in the case of a wide chest would impart a very unnatural “double cone” appearance. For women with a mid-range chest-wall diameter the choice is one of partiality. That is, the decision has to be made between projection, and cleavage. This is because lower profile implants with a wide base will naturally fill up the inner, otherwise known as the medial breast, and produce cleavage. Finally, it is mostly the anecdotal opinion of some authorities that full or high profile implants tend to generate less rippling.
What is wound dehiscence after breast augmentation repair or revision surgery, what are the consequences, and what is the treatment?
Wound dehiscence is a disruption of the incision used to access the breast implant pocket in breast enlargement. It may be due to infection, impaired healing, or post-operative trauma. If due to infection, antibiotic therapy or even removal may be necessary as outlined in the question on infection. If due to impaired healing, the precise factor(s) must be identified and addressed, though the breast implant needs may be salvaged. If Caused by trauma, and no breast implant exposure is noted, either primary, or delayed wound closure may be used.
Should my physician use a shaped or round implant in performing my breast augmentation repair?
This decision depends as much on what the surgeon can accomplish using either the round or the anatomically contoured implants as on which type of implants to use.
Assuming the breast pockets are dissected in to the inferior or lowermost extent in the exact same manner, the shaped implants will impart more fullness at the top, and provide a more natural, gently curving slope to the augmented breast. For this to happen, however, the lowermost portion of the dissection or pocket must be the same no matter which mammary implants you use. If a surgeon habitually dissects low enough inferiorly to drop round breast prosthesis low enough that upper pole fullness is lost, that surgeon will likely favor the teardrop shaped, or anatomically shaped implant to compensate for that dissection. For anatomically shaped or contoured implants to do what they were intended for, pocket dissection has to extremely precise. This is because contoured breast implants are not symmetric; they have a top and bottom. If the subglandular or submuscular pockets are too wide, the shaped implants can shift or even flip, imparting asymmetry and even worse an unnatural shape to the augmented breast. Even though shaped mammary implants are textured, this is still no guarantee against malposition. Finally using the round type of breast implants can impart more medial or inner breast fullness. This significantly improves cleavage, because more fill volume winds up toward the more central part of the breast.
What is an axillary incision in repair breast enlargement surgery?
Placing the incision in the axilla (underarm) allows for a smaller incision. It is useful when there is not much breast tissue present to begin with, and thus no prominent breast fold, and when the nipple-areola complex is small. It is not as well concealed as the periareolar incision, gives less control in terms of the ability to feel around the breast pockets in assuring symmetry, and placement of silicone breast implants will necessitate a larger incision which may be less pleasing.
Is bleeding common after breast augmentation revision surgery, what are the consequences, and what is the treatment?
Bleeding is not a common complication after breast augmentation surgery; however, it may lead to problems if not detected in a timely fashion. This applies more to acute hemorrhage immediately after breast enlargement surgery. The affected side would become considerably larger than the non-bleeding side, would possibly turn pale and purplish, and the resulting tension would potentially threaten the viability of the overlying skin. The volume of blood that a breast pocket could potentially accumulate is not large enough to be life-threatening unless combined with certain other conditions. The treatment for acute postoperative bleeding after breast augmentation surgery is an immediate return to the operating room, release of the collected blood, a search for the offending vessel, and control of the bleeder. A hematoma may form after breast augmentation surgery in the case of a slow bleed, not detected immediately, or with subsequent trauma, days, weeks, or months after the initial operation for breast enlargement. The result would be swelling, discoloration, and pain. If large, as judged by the operating surgeon, the collection would need drainage. Smaller hematomas can be absorbed by the body.
Will there be breast skin and breast tissue changes after silicone gel-filled breast implant use in breast augmentation revision surgery?
Breast implantation stretches the skin envelope of the breast. This can accentuate stretch marks. Over time, breast skin can thin, revealing breast implant irregularities and fold flaws. The additional weight can potentially lead to an increase in breast drooping, and necessitate breast lifting or tightening.
What other points should I consider when chosing silicone gel-filled breast implants to be used in my breast enlargement revision surgery?
In addition to all of the possible complications, and unplanned additional surgeries you should also consider the possibility of health insurance premium increases. It may also be a good idea to invest inCosmetAssure®. This is an independent insurance company that offers coverage for patient treatment in cases of post-surgical complications related to cosmetic surgery for a one-time premium.
Who would be considered a good candidate for revision or repair of breast enlargement or breast augmentation surgery?
There are two important factors a board certified plastic surgeon will take into account when determining whether or not a patient is an appropriate candidate for revision breast augmentation surgery. The first consideration is a clear definition of the potential patient’s concerns, and clearly set expectations. As an example, a woman may be unhappy with the position of one breast fold with respect to another, with asymmetry of her breasts after enlargement, with firmness, etc. Vague desires that are open to interpretation can lead to remarkable dissatisfaction, regardless of how well the surgeon thinks the operation was performed.. The second consideration is the client’s psychological and otherwise motivation for desiring repair of a previous breast enlargement. The patient who is emotionally stable, secure, has thought the process through, is aware of possible drawbacks, and has set realistic goals is likely to be very satisfied with the outcome of revisionary or secondary breast enlargement surgery. The insecure client who believes that repair of their breast augmentation is the next new key to changing their life, who is pushed to repair by a need to satisfy a partner, or who decides to undergo repairative breast enlargement surgery on the spur of the moment will be highly likely to be displeased by any post-surgical outcome, no matter how outstanding. The third consideration is the patient’s overall health condition. It goes without saying that high-risk patients with multiple medical issues should not undergo elective cosmetic surgery.
What are the polyurethane breakdown products?
The MHRA publishes a review of the products.
“Morag E. SaundersJeremy J. B. TinklerMedical Devices Agency
July 2001
REVIEW OF THE BIOLOGICAL SAFETY OF POLYURETHANE-COATED BREAST IMPLANTS
SummaryPolyurethane-coated breast implants were the subject of two advisory notices by the
Medical Devices Agency, in 1994 and 1996. These highlighted reports from theliterature which suggested that degradation of the polyurethane coating occurred overtime producing the potentially carcinogenic 2,4-toluenediamine (2,4-TDA). The UKDepartment of Health’s Committee on Carcinogenicity (COC) had advised, in 1991and 1994, that the breakdown of the polyurethane over a period of several years,leading to the presence of 2,4-TDA in the tissues surrounding breast implants, gave
rise to a small, unquantifiable, carcinogenic risk. Specifically, the COC concludedthat:
2,4-toluenediamine (2,4-TDA) should be regarded as a probable genotoxic carcinogen;
there is evidence that small amounts of 2,4-TDA can be released from polyurethane foam in vitro and in vivo;
direct evidence exists that 2,4-TDA is released from polyurethane coated breast implants in vivo in women. This leads to the presence of 2,4-TDA in the tissues surrounding the implant. These tissues can metabolise 2,4- TDA and there is therefore a potential carcinogenic risk from this release;
information on the breakdown of implanted polyurethane foam in human tissues suggests that breakdown occurs over a period of several years;
there were no data on the possible release of harmful substances other than 2,4-TDA from implanted polyurethane foam either in vitro or in vivo.
A review of more recent data has now been carried out by MDA. This has revealed the following new findings:
DNA damage has been shown in animals following oral administration of2,4-TDA, but not following implantation of polyurethane foam disks;
2,4-TDA has been identified in the urine, wound drainage fluid and, for up to 2 years after implantation, bound to plasma components, of women with polyurethane-coated breast implants.
Recent evidence is thus consistent with the conclusions reached by the COC in 1991 and 1994.
The results of studies on the rate of degradation have been used as a basis for quantitative risk assessments. Three separate estimates concluded that the lifetime cancer risk resulting from the implantation of polyurethane-coated breast implants to be in the region of 1 in a million and thus of minimal toxicological risk. However, risk assessments of this sort are not consistent with UK scientific policy that exposure to genotoxic carcinogens should be eliminated wherever possible and, where it cannot be avoided, should be reduced to a level as low as is reasonably practicable.
Moreover, the European Medical Devices Directive requires that any residual risks be outweighed by benefit to the patient. Thus, in assessing the suitability of polyurethane-coated breast implants, it is necessary to take into consideration both the avoidability of the risk and any benefit arising from the use of the product.
The principle benefit claimed for this type of implant, in comparison with other breast implants, is a reduction in the rate of capsular contracture. While there is evidence to support this claim, uncertainties exist over the degree and persistence of the benefit and it is not possible to conclude that the benefits claimed for polyurethane-coated breast implants can only be achieved through the use of this particular coating material.
It is concluded that there remains insufficient evidence for benefits arising exclusively from the use of the polyurethane coating applied to these breast implants to justify exposure to the carcinogenic risk, albeit very small, associated with its breakdown in situ.
Policy in the UK should therefore remain that polyurethane-coated breast implants should not be implanted, but that the removal existing implants is not indicated.
BackgroundA polyurethane foam coating was developed for silicone gel breast implants in the1980s because it was felt that the open texture of the foam would modify thealignment of cells in the fibrous capsule surrounding an implant and thus reduce theincidence of capsular contracture. Two models of polyurethane-coated breastimplants (Même and Replicon) were developed in the USA, using a commerciallyavailable polyesterurethane foam. Following concerns that degradation of thisparticular polyurethane could lead to the release of a carcinogenic breakdown product, the original American manufacturer voluntarily withdrew the products from the market in 1991. Similar products were subsequently re-introduced to the Europeanmarket by other manufacturers but very few were used in this country before allmanufacturers ceased supply to the UK at the instigation of the Department of Healthin 1993.
As part of an on going process, the Medical Devices Agency (MDA) continues tomonitor the safety of materials used in the manufacture of breast implants available in the UK. Recent controversy over silicone-filled implants and the events surrounding Trilucent. breast implants, prompted MDA to review available data on a number of these product types including two hydrogel-filled breast implants. Although polyesterurethane-coated implants, are not currently available in the UK. Themanufacturer has indicated a wish to place these implants back on the UK market,pointing out that two extant Safety Notices published by MDA warning against theiruse represent a technical barrier to trade which, if unjustified, would run counter to European single market legislation.
Assessments in 1991 and 1994Two previous reviews of the carcenogenicity of polyurethane-coated breast implantshave been carried out by the MDA in 1991 and 1994 and both reports were submittedto the Committee on Carcinogenicity (COC) for review. In 1991, concerns wereraised over the degradation potential for of polyurethane and the release of 2,4-and2,6-toluinediamine (TDA) and the fact that 2,4-TDA is a known rodent carcinogenwhich has been shown to be released from polyesterurethane in vitro followinghydrolysis.
The 1994 report strengthened the view that 2,4-toluenediamine (2,4-TDA) isproduced from the degradation of polyurethane breast implants in vitro. Furtherevidence was presented which showed that 2,4-TDA is produced from the degradationof polyurethane breast implants in vivo. However, there was insufficient dataavailable at the time to comment on the rate of degradation of polyurethane.
The 1994 report was submitted to the COC, who concluded that 2,4-TDA should beregarded as a probable genotoxic carcinogen when administered orally. Theyconsidered that direct evidence existed that 2,4-TDA was released form polyurethane-coated breast implants in vivo in women which led to the presence of 2,4-TDA in thetissues surrounding the implant. These tissues could metabolise 2,4-TDA and therewas therefore a potential carcinogenic risk from this release. The information on the breakdown of implanted polyurethane foam in humans suggested that breakdownoccurred over a period of several years. Overall, the COC considered thatpolyurethane was not a suitable material for implantation as it presented a small, but unquantifiable carcinogenic risk.
The conclusions of the COC were published in the 1991 and 1994 Annual Reports ofthe Committees on Toxicity, Mutagenicity, Carcinogenicity of Chemicals in Food,Consumer products and the Environment. In addition, the implications of the findingslead to the issue of two Safety Notices (SAB(94)39, September 1994 and SN 9620,July 1996) by the MDA.
Situation at PresentAt present polyurethane-coated breast implants are CE-marked (through the GermanNotified Body, MDC) and distributed in Germany, Italy and Spain. In the light of the1994 COC opinion and the two Safety Notices, the manufacturer (Polytech Silimed)has indicated that the product will not be marketed in the UK but has pointed out that this situation runs counter to European free market legislation. The manufacturer claims that new evidence is now available in support of the safety of polyurethane-covered breast implants. In particular, that the amount of 2,4-TDA released does not pose a carcinogenic risk. Polytech Silimed is keen to have the situation regarding the UK market reviewed and has supplied the MDA with further information in support of their claim that there is no risk of cancer from these breast implants. The company pointed out that this information has become available since the MDA review of 1994 and that it has not so far been taken into account. This paper presents a review of the literature supplied by Polymed Silitech in support of their claim that these implants do not pose a safety threat. Other relevant publications obtained through a literature search carried out by MDA are also considered.
Structure of Polyurethane
The polyurethane used in the manufacture of breast implants is a polyesterurethanemanufactured from polyethylene glycol adipate (PEGA) and toluene diisocyanate(TDI). The latter comprises of a mixture of 2,4-TDI and 2,6-TDI isomers in a 4:1ratio. TDI is unstable in aqueous environments and reacts to form the equivalenttoluenediamine (TDA) isomers.
All polyurethanes are to a greater or lesser degree susceptible to hydrolysis of theurethane bond with production of low molecular weight fragments. The polyesterurethanes are also susceptible to hydrolysis of the ester linkages with aninitial production of low molecular weight fragments and, ultimately, the componentsof the polyester diol. The ether linkages in polyetherurethanes are more resistant to hydrolytic cleavage and are essentially hydrolytically stable.
New Data In Vitro StudiesA study conducted by LPU (1996) on the behalf of Polytech Silimed investigated therelease of TDA under physiological conditions. Samples of the polyurethane coatingof breast implants were extracted over a period of 30 days at 37oC both in thepresence and absence of papain. The extraction medium was exchanged every twodays for 60 days and the levels of 2,4-TDA and 2,6-TDA measured in each solution.Both sterilised and non-sterilised material was tested for the release of 2,4-TDA and 2,6-TDA. Sterile and non-sterile samples were also incubated in papain for 14 dayswithout changing the papain solution and in saline for 30 and 60 days only.
No detectable levels of either 2,4-TDA or 2,6-TDA were found in the extracts fromeither the sterile or non-sterile samples treated with papain when the extract solution was changed every two days over the 60-day incubation period. The quantity of TDA released within two days was considered to be below the analytical limit for 2,4- and 2,6-TDA. In samples incubated in papain over 14 days with no change of solution or in a sodium chloride solution, over 30 and 60 days, up to 245 ppb of 2,4-TDA and 246 ppb of 2,6-TDA were released from both the sterile and non-sterile material.
From their data based on a total weight of 8 g of polyurethane incubated with 0.9%salt solution for 60 days, a total quantity of approximately 1.5 µg/ml 2,4-TDA and 2.3 µg/ml 2,6-TDA were recovered. Extrapolated over a year, this gives a total quantity released of 9 µg/ml 2,4-TDA and 13.8 µg/ml 2,6 TDA. Linear release rates were assumed making the release of 1-2 µg/ml 2,4-TDA equivalent to approximately 0.02-0.03 µg/ml per day. The authors compared these values with the dose levels used in the studies carried out by the National Cancer Institute where rats and mice were fed substantially higher dose levels. They suggested that the release rates determined for 2,4-TDA were relatively low and this may be the reason why no effects were seen in carcinogenicity and genotoxicity studies where polyurethane foam or extracts of foam were used in the relevant test systems. This suggestion is of limited validity, since a quantitative comparison of in vitro and in vivo studies is of questionable relevance.
The findings of the Polytech Silimed/LPU Report are in marked contrast to those ofSzycher and Siciliano (1991) who previously examined the hydrolysis of washedpolyurethane foam over 30 days in an aqueous solution of papain. They found that2,4-TDA and 2,6-TDA were released only within the first 4 days of incubation.
The in vitro biodegradation of polyurethane foam was also discussed by Lubbers(1997) who reviewed several reports where separation of the polyurethane coating ofimplants was observed. The chemical hydrolysis and/or chemical breakdown of thepolyurethane coating were considered to be of more importance. Many of the studiesreferred to in this text were discussed in the 1994 MDA report. In summary, TDAwas released from foam samples incubated in NaOH at 37oC. No TDA was extractedfrom samples incubated in normal saline but trace quantities of 2, 4-dimethyl-6-t-butyphenol were washed out after incubation in methanol (Daka and Chawla 1993).Battich and Williams (1989) also detected TDA after incubation in NaOH. Amin et al. (1993) were unable to extract TDAs from foam incubated in either methyl tert-butyl ether or aqueous buffer extracts. Only incubation of foam or foam extracts ineither strong mineral acid or base resulted in TDA release. On the other hand, Benoit (1993) showed that polyurethane foam released 2,6-TDA, 2,4- and 2,6-TDI andtoluene isocyanate (TIA) following incubation in Ringer’s solution at 37 oC forperiods of 6 to 36 days. The investigations of Szycher and Sicillano (1991) whofound that 2,4-TDA was released only within in the first four days after enzymaticdigestion were also discussed. As mentioned above, this finding is in contrast to those found in the LPU investigation (see above). The degradation of polyurethane-coated implants has also been reported by Sinclair et al. (1993) and by Brorson et al. (1991).
The report by Lubbers concluded that ‘although several studies showed that thatbiodegradation of polyurethane occurred and that in some cases TDA had beendetected in urine, there was no clinical or epidemiological proof that breast implants cause cancer in any way’. The risk benefit analysis concluded that ‘the great advantage of the polyurethane-coated breast implants is the low frequency of capsular contracture and that the negative side effects do not exceed those of other breast implants’.
An extensive search of the literature found few recent reports (post 1994) directlyrelated to the breakdown of polyurethane used to coat breast implants and the release of 2,4-TDA. However a number of studies on the breakdown of polyurethane foam ingeneral were found. In vitro biodegradation studies on polyesterurethane synthesisedwith radio-labelled toluene diisocyanate (TDI) (Santerre et al. 1994) were carried out using cholesterol esterase (CE) and Horseradish peroxidase (HRP). Highperformance liquid chromatography (HPLC) analysis of the products showed that thebulk of the TDI remained covalently bound within the cleaved chain segments of theoriginal polymer and not released as pure toluene diamine. Further studies (Wang etal. 1997) with poly(ester)urea-urethanes synthesised with radio-labelled TDI andincubated with CE generated a number of biodegradation products. TDA, was notdetected by chromatographic separation although two products were identified whichwere TDA derivatives (secondary aromatic diamine) substituted with end units of thepolyester segment at N and N’ positions of TDA. The authors suggest that there couldbe a stabilisation of the urethane and urea linkages within the TDI segments of thepolyurethanes.
Summary of In Vitro DataFor the most part, the in vitro studies support the view that small amounts of 2,4-TDA are released from polyurethane following various treatments. In many cases, the amounts of 2,4-TDA released were low. In two of the more recent studies, TDA wasnot detected.
In Vivo StudiesLuu et al. (1998) described a physiological based pharmacokinetic model to simulatethe fate of low dose exposure of 2,4-TDA leached from polyurethane foam-coveredimplants. They used their model to assess the potential health risk to humans. Ratswere given 0.52 mg/kg 2,4-TDA by iv bolus, orally or 0.021 g by implantation. Theconcentration of 2,4-TDA was measured in five tissue compartments (kidney, gastro-intestinal tract, liver, fat, muscle) as well as in plasma and urine. Based on apolyurethane foam mass of 4.87 g the results were extrapolated to determine valuesfor implant simulation in rats and humans and low dose ingestion in the rat. Fromtheir results the authors predicted an excess lifetime average daily dose in humans of 11.93 x 10-6 mg/kg/day with an increased risk of breast (and liver) cancer of 1 in 400,000.
The results of the Luu study have been criticised for over-estimation of the dose ofpolyurethane, incorrect measurement of the breakdown products and the scalingfactor used (Kulig 1998). Their estimate of risk (1 in 400,000) was considered to begreatly over estimated. However correcting for the greater weight of the implant used in the study (4.87 g cf 2.7 g), the estimated exposure value is approximately equal to that of the FDA (1991) and Hester et al. (1997) (i.e. 1 in 1,000,000).
Bradley at al. (1994a) investigated the subchronic immunotoxicological potential ofthe principal constituents of silicone (fluid, gel and elastomer) breast implants and of polyurethane. The silicone fluid and gel were injected subcutaneously and disks(6mm) of the silicone elastomer and the polyurethane were implanted subcutaneouslyinto B6C3F1 mice. Immunotoxicological and host resistance studies were carried outafter 10 days. They found no treatment-related deaths or overt signs of toxicity.None of the tested materials had notable effects on body or organ weights,erythrocytes or leukocytes in the blood or blood chemistries. The tested silicones did not alter the distribution of B cells and T cells in the spleen, but some perturbations in the levels of CD4+CD8+ and CD4-CD8- T cells were seen in the polyurethane-treated mice. The antibody response to sheep erythrocytes was not markedly altered nor were the proliferative responses to concanavalin A, phytohemagglutinin, lipopolysaccharide or allogeneic cells. Reticuloendothelial function was normal but polyurethane evoked an enhance phagocytosis of Covaspheres by adherent peritoneal cells. Natural killer cell activity and serum complement were not altered. All of the materials showed some protection to a challenge with Listeria monocytogenes that killed 40–58% of vehicle control mice. Host resistance to Streptococcus pneumoniae or the B16F10 tumour was not affected. The results suggested perturbation of T cell differentiation in mice implanted with a polyurethane disk.
In a similar long-term, 180 day exposure study Bradley et al. 1994b showed the onlyconsistent effect of exposure to silicone materials or polyurethane was a modestdepression of natural killer cell activity. No indication is given in either study as to the anticipated levels of polyurethane exposure or of the form of the polyurethane.
Delcos et al. (1995) assessed the extent of DNA damage in rats fed 2,4-TDA underconditions that result in tumour induction and in rats implanted with Microthanepolyesterurethane foam (67 mg/kg or 267 mg/kg). Time and dose dependent adducts were found in the DNA from the liver and mammary glands of rats fed 10, 40, 80 or180 ppm 2,4-TDA for up to 6 weeks. In the rats fed 40 or 180 ppm 2,4-TDA, DNAadducts were detectable in liver and mammary tissue for 26 – 43 weeks after thetermination of treatment. No adducts were detected in T lymphocytes isolated fromthe spleens of rats fed 40 or 180 ppm 2,4-TDA nor was there an increase in mutationsat the hprt locus in these lymphocytes. This indicates the potential for 2,4-TDA tocause genotoxic effects in vivo.
In the implantation groups, no DNA damage was observed in the liver, mammaryglands or T-lymphocytes of rats up to 42 weeks post implantation (67 mg/kg) or in the liver and mammary glands of rat implanted with 267 mg/kg polyesterurethane foam.
The authors calculated that a 50 kg woman with 2 implants would receive 54 mgpolyurethane foam per kg body weight. They were unable to detect 2,4-TDA-relateddamage in rats treated with 67 or 268 mg foam per kg body weight and pointed outthat rat bioassay data indicated that a dose of 4.7 mg 2,4-TDA/kg/day was sufficientto induce tumours in rats.
While the results showed the development of DNA adducts as a result of systemicexposure in rats fed 2,4-TDA, the tissues immediately surrounding implants did notappear to have been analysed for adducts. The levels of 2,4-TDA used in the studycorresponded to time-weighted average doses used to demonstrate tumourigenicity(US Department of Health, Education and Welfare bioassay for 2,4-TDA). While2,4-TDA was capable of damaging DNA, the authors concluded that the polyurethanefoam implants presented a minimal risk of genotoxicity through release andsubsequent metabolic activation of 2,4-TDA.
Summary of In Vivo DataRecent in vivo data suggest that polyurethane does not cause immunoreactivity in a rat model. Of greater importance is the identification of DNA adducts in the liver and mammary tissues of rats fed 2,4-TDA, although no DNA damage was observed in ratsimplanted with ployurethane foam disks for 42 weeks.
Clinical StudiesA number of clinical studies relevant to the safety of polyesterurethane breastimplants have been carried out. These have included studies on the levels of 2,4-TDAreleased from the polyesterurethane coating, systemic effects relating to implantation and the occurrence of capsular contraction.
Exposure AssessmentAt the request of the FDA, Bristol-Myers Squibb carried out a study to determinewhether the polyurethane foam covering silicone breast implants could release theknown animal carcinogen 2,4-TDA. The results of this study were published in areport by Hester et al. (1997). The initial results were described in an interim report by Ford et al. (1993) and referred to in paragraph 48 of the 1994 MDA report which was reviewed by COC.
The urine and blood serum of 61 women with polyurethane-covered implants and 61women without implants were tested for the presence of TDA. The study reportedthat tiny amounts (parts per trillion) of TDA were found in the urine of 80% of thewomen in the study with breast implants. Smaller amounts of free TDA were foundin the urine of 13% of the women without implants. No free TDA was found in theblood of the women with polyurethane implants.
As noted in the 1994 MDA report, the study showed that 2,4 TDA is released frompolyurethane foam in vivo. Hester et al. (1997) and the FDA used the results of thisstudy to estimate the lifetime average daily dose of 5.06 x 10-6 mg per kg per dayTDA resulting from the breakdown of the foam. To this they applied the cancerpotency factor (0.21 mg/kg/day) previously used by the FDA which gave an estimatefor the upper limit of excess lifetime risk resulting from the implants of 1.1 x 10-6. This represents a theoretical lifetime risk of about 1 in 1 million. The report concluded that while the polyurethane foam coating of the implants degrades, the risk of cancer in humans from exposure to TDA is negligible.
Sepia et al. (1995) also measured levels of TDA in vivo in six patients implanted with polyurethane-covered breast implants. They monitored levels of TDA in blood, urine and wound drainage. They reported that following acid hydrolysis of urine samples elevated amounts of TDAs were found in post-operative test samples. High levels of TDAs were found in the wound drainage samples which showed a sharp drop over the first four days. The levels of 2,4-TDA and 2,6-TDA in plasma rose after an initial lag period of 20-30 days, remaining elevated for up to 2 years. TDA released from total plasma proteins under acid conditions were not released under mild base treatment. When the plasma proteins were precipitated in ethanol, redissolved and treated with a mild base, TDA was still not released. Protein was however released after acid hydrolysis of the precipitated samples. The levels of TDA released from globulin and albumin fractions subjected to acid hydrolyis both in the presence and absence of precipitation in ethanol was similar.
This paper gives further evidence of the presence of polyurethane degradationproducts in the urine and plasma of implanted patients for a period of up to two years. The TDA-derivatives appeared to be covalently bound to plasma proteins. Thelifetime risk of cancer was assessed on the basis of protein adducts. A post-operative level of 4.4 ng 2,4-TDA/ml plasma was calculated which is equivalent to 10.56 µg 2,4-TDA in a 60 kg subject with a plasma volume of 2,400 ml. The kinetics of globulin adducts were considered to be the same as those for plasma and an adduct level of 352 ng was calculated which was used to calculate a daily dose of 70.4 µg2,4-TDA. This figure was compared with the dose used in the risk assessment carriedout by the Canadian Medical Association (1991) who calculated that a 236 ng (0.236µg) TDA would be released per day from two implants containing 2.7 g polyurethane.
Hester et al. (1997) measured free TDA in the serum and urine of implant patientswhile Sepia et al. (1995) measured the level of bound TDA. The results are indicative of continued degradation of the polyurethane coating on the implants. Both papersconcluded that no free 2,4-TDA was present in the circulation. The estimated valueor annual exposure calculated from available data and averaged over 6 yearsassuming linear degradation suggested a value in the range of 28 – 41.4 mg. These are similar to the figures reported by Sepia (1995).
De Lorenzi et al. (1995) reported on an HPLC method for the determination ofurinary content of 2,4- and 2,6-TDAs in patients with polyurethane breast implants.Urine was collected from 6 patients at several time points over a period of 8 months to 8 years. The urine was analysed using reverse phase HPLC with fluorescencedetection for the simultaneous determination of 2,4- and 2,6-TDAs followingliquid/liquid extraction and derivitisation. Most of the samples analysed werenegative for TDA with the exception of the urine from one patient in which adetectable level of 2,6-TDA was found. The limit of sensitivity of the technique wasgiven as 15 ng/ml urine.
Tissue Response and Clinical OutcomeOther studies have been reported which describe the histological reactions associatedwith implantation of polyurethane-coated breast implants. Handel et al. (1995)reported on a follow-up study of patients which involved 1655 breast implants over aperiod of 15 years. The study was designed in order to overcome the inconsistenciesand incomplete data records that existed for implant patients. A comparison ofvarious breast implant types, including polyurethane, was made and a number ofcriteria were assessed (e.g. capsular contracture, surface texture, implant position, skin wrinkling, skin rash) by various statistical methods. In the short term, the risk of capsular contracture was similar for textured and polyurethane-covered implants but less for smooth implants. On the other hand, the polyurethane-covered implants significantly reduced capsular contracture over the entire follow-up period. However, a higher incidence of skin rash and to a lesser extent skin wrinkling was associated with these implants compared the majority of the other implant types (Siltex, smooth implants). Biocel implants showed a much greater frequency of skin wrinkling than the polyurethane-covered implants. The polyurethane-covered implants were shown to be superior to both smooth and textured silicone implants in terms of reducing the risk of capsular contracture.
The literature review carried out by Lübbers (1997) and supplied by Polytech Silimedset out to clarify whether the aim to decrease capsular contracture without increasing other side effects had been achieved with polyurethane foam-covered breast implants. The review compared the risk of infection in relation various types of implants (smooth, textured, polyurethane-coated). From the papers reviewed, they concluded that the risk of infection was no greater with polyurethane-covered implants. However, some reports linked polyurethane-covered implants to the appearance of an inflammatory response including a rash. A number of studies, both in rats and humans, were discussed and it was concluded that use of polyurethane-covered implants reduced the occurrence of capsular contracture compared to other implant types. All of the literature reviewed pre-dated the MDA report of 1994.
Vázquez (1999) reported on a series of 407 patients with polyurethane-coveredimplants carried out over a period of 10 years. Of the 811 single implants inserted, 24 were of the Même National White type, 6 were the Replicon surgitex model and 781 were Silimed. Only 0.49 % of the total implants inserted showed capsularcontracture.
Light microscopic analysis showed the capsule could be divided into five layersaccording to histological composition. The inner layers were composed ofmacrophages, foreign body giant cells and inflammatory cells and the outer layersconsisted of fibrous connective tissue. The capsules showed a lower concentration ofcollagenous fibres compared to other types of implants. Enzymatic degradation ofcapsules showed the presence of polyurethane remnants confirming that thepolyurethane is digested in the capsule. Light microscopic examination andimmunological typing of the capsule showed the presence of a chronic inflammatorycell infiltrate.
A number of papers report associations between polyurethane-coated breast implantsand cellular reactions. Luke et al. (1997) studied capsular tissue from 86 cases inorder to characterise the relationship between capsular findings and the type ofimplant used. Cellular reactions were associated with all implant types examined butwere most prominent in capsules associated with polyurethane-coated implants. Thecellular reaction consisted of vacuolated macrophages, chronic granulomatousinflammation and large numbers of multinucleated giant cells, some containingasteroid bodies. In addition, particulate material identified as polyurethane by FTIR microscopy was found in all cases where polyurethane-coated devices wereimplanted. A pseudoepithelium was seen at the inner capsular surface (synovialmetaplasia). Immunohistochemical studies suggested the pseudoepithelia were ofmacrophage/histiocytic origin.
In a single case study, Raso and Greene (1995) also reported on synovial metaplasiain the periprosthetic capsule. The synovial metaplasia was considered to develop inresponse to the polyurethane-coated implant and was implicated in reducing capsularcontracture and increasing host acceptance of implantable biomaterials. It wassuggested that a synovium-lined periprosthetic capsule lubricates the luminal cavityand could theoretically reduce friction at the prostheses-capsular interface.
A report by Wang et al. (1998) describes the development of two cases of latehaematomas after breast reconstruction with polyurethane-coated implants. The exactorigin of this adverse effect was not explained, but may have been due to the highlyvascular inflammatory response associated with the polyurethane coating of the breast implants. It was suggested that the extensive inflammatory and foreign body reaction seen with polyurethane-coated implants prevents fibroblasts being laid down in theregular fashion as seen with other implants, giving a softer capsule less likely tocontract. However, disruption of the vascular component of the reaction could havelead to the late bleeding observed.
A comparison of systemic and rheumatic disease manifestations was reported byBulpitt et al. (1998). From 250 patient cases, they randomly chose 25 patients withpolyurethane-coated breast implants and compared these with 25 patients with non-polyurethane-coated breast implants. Patients with the polyurethane-coated breastimplants showed a shorter time between implantation and symptom onset. A possibleassociation with a higher incidence of mucocutaneous lesions (rash, photosensitivity, oral ulcer) was suggested.
In a population-based case-control study, Brinton et al. (1996) showed the relativerisk of breast cancer was reduced with a prior implant when compared to the controls. This rate persisted with increasing interval since surgery and was lower for bothlocalised and distant tumours.
Summary of Clinical StudiesClinical studies have indicated the presence of 2,4-TDA in the urine, wound drainagefluid and blood (bound to plasma components). In one study, 2,4-TDA was detectedin the urine and plasma for up to 2 years post-implantation, confirming continuedpolyurethane degradation.
A reduction in capsular contracture with polyurethane-coated implants compared toother types of implant has been reported. However other contra-indications have beenreported including, skin rash and skin wrinkling, chronic inflammatory reactionsassociated with the capsule and the presence of granulomatous reactions composed ofcells containing particles of polyurethane indicating breakdown of the implants. Theinflamatory changes may be related to the effect on capsular contracture.
DiscussionThe information supplied by Polytech Silimed has been reviewed along with otherrelevant data from the literature. The majority of the data supplied by PolytechSilimed had previously been considered by the UK Medical Devices Agency (MDA)and was discussed in their 1994 report to the UK Government’s expert advisorygroup, the Committee on Carcinogenicity (COC). In particular, the data reported byHester et al (1997) had already been considered by the COC and the literature surveycarried out by Lübbers (1997) included many of the studies previously reviewed.MDA carried out an extensive literature search and a number of additional paperswere identified and reviewed that were relevant to an assessment of toxicity orexposure in relation to polyurethane-coated breast implants.
The nature of the degradation products depends on the original formulation of thepolyurethane foam. It has been established, from in vitro studies carried out undervarious conditions, that 2,4-TDA is released from the particular polyurethane foamchosen for use in the Polytech Silimed implant. The release of 2,4-TDA was found tobe dependent on the extract medium used although the timing of release was notalways consistent.
While it is widely agreed that 2,4-TDA is released from polyurethane implants invivo, not all workers have been able to detect the presence of TDAs. Two papersconcluded that no free 2,4-TDA was present in the circulation and one study was ableto detect TDAs in the urine of only one out of six patients with polyurethane breastimplants. The fact remains, however 2,4-TDA has been detected in the urine andbound to plasma components in implanted women and the release of 2,4-TDA fromthe breakdown of polyurethane foam has been demonstrated under in vitro, in vivoand clinical conditions. This evidence for the breakdown of the polyurethane foamcovering the implants cannot be ignored.
The Critical Issues Are:
Whether the levels present in women with polyurethane implants represent asignificant toxicological risk, and
Whether the residual risk is acceptable when considered in relation to anybenefits to patients arising from the presence of the polyesterurethane foam.
MDA policy in addressing these questions arises from guidance provided by the UKGovernment’s expert committees on Carcinogenicity (COC) and Mutagenicity(COM), the European Medical Devices Directive (93/42/EEC), and the internationalstandards ISO/DIS 10993-17 (1999) and EN ISO 14971 (2000).
Following their review of the 1994 MDA report, the COC concluded that 2,4-TDAshould be regarded as a possible human carcinogen There was evidence that smallamounts of 2,4-TDA could be released from polyurethane in vitro and indirectevidence of breakdown in vivo in rats. At the time, the quantity and identity of thebreakdown products had not been established and the possible effects on local tissues were not known. In addition, no information on the breakdown of implantedpolyurethane foam in human tissues was available. The more recent observation ofDNA adducts in the liver and mammary gland in rats fed 2,4-TDA confirms that 2,4 TDA is genotoxic in vivo. Evidence of the presence of 2,4-TDA in women withpolyurethane breast implants is also now available.
While it is accepted that the amount of 2,4-TDA released from an implant, and thusthe risk, decreases over time, the absence of data on local tissue levels of 2,4-TDAwas of particular concern to the COC, since these tissues were felt to be at greatest risk. More recently reported changes in the structure of the polyurethane coating and the presence of fragments of the foam support the COC’s view, based on the 1994MDA report, that there is complete break down of the polyurethane. No further datacould be found in the literature from which to estimate the rate of degradation of the polyurethane coating in vivo or which address local tissue levels of 2,4-TDA after implantation.
While no reports exist of tumour development as a direct result of the implantation of polyurethane-coated breast implants, this cannot be used to discount the risk ofcarcinogenesis, as any tumours might arise several years after implantation. It isunlikely that data on human cancer incidence will ever provide any useful insight into the carcinogenic risk arising from polyurethane-coated breast implants.
Three quantitative estimates of the lifetime risk of cancer arising as a result of the implantation of polyurethane coated breast implants have been reported. Theseassessments reach a consensus that the risk is in the region of 1:1,000,000. A number of risk assessment models characterise a risk of this order as negligible and a case can be made for considering any risk of this magnitude to be tolerable. The magnitude of a risk, however, is not the only factor that needs to be taken into account in determining its acceptability. In order to be confident that a risk is “so low that it is not worth bothering about”, it is also necessary to consider the nature of the hazard, the balance of risks and benefits and whether the risk is avoidable or undertaken voluntarily (EN ISO 14971).
In assessing the nature of the hazard, the critical concern is that we are dealing with exposure to a genotoxic carcinogen. A precautionary approach is adopted in public policy in the UK for such risks (COM, 2001). It is considered prudent to assume that there is a linear, non-threshold dose response to in vivo mutagens. For risk management purposes in the UK, it is assumed that any exposure to such chemicals results in some damage to DNA and thus an increased risk of mutation leading, inturn, to an increased risk, albeit possibly very small, of adverse health effects. The COM recognises specific exceptions to this rule, in situations where thresholds can be identified for mutagenicity, but considerable mechanistic data are available to justify these exceptions. No such data are available with respect to 2,4-TDA.
The consequence of scientific policy in the UK is that a tolerable exposure limit to a genotoxic carcinogen or in vivo mutagen cannot be established. Instead, a riskmanagement policy is adopted whereby exposure to such compounds is eliminatedwherever feasible and, where it cannot be eliminated, is reduced to a level “as low as reasonably practicable” (ALARP). In the absence of convincing reassuring data, the observation that DNA adducts are formed in vivo on exposure to 2,4-TDA, requiresthat this approach must be adopted in the UK in the case of polyurethane-coatedbreast implants.
In many other countries, however, including the USA, a quantitative risk estimate isconsidered an acceptable basis for the risk assessment of genotoxic carcinogens.Indeed, ISO/DIS 10993-17 allows either approach to be selected, based on theprevailing regulatory scientific policy. A strong case can thus be made forcompliance of a purely quantitative risk assessment with ISO/DIS 10993-17.Although this draft standard is intended to become a harmonised European standard,it cannot be assumed that the quantitative risk assessment option will remain available to those who wish to use the standard to claim compliance with the Medical Device Directive. This is an issue that has yet to be considered by the relevant European Standards Technical Committee and the European Commission.
EN ISO 14971 and ISO/DIS 10993-17 require that any residual risk, unless it can beclassified as negligible (i.e. “so low that it is not worth bothering about”), must be outweighed by the benefit obtained from exposure to it. The risk arising fromexposure to an in vivo mutagen cannot be considered by the UK Department of Healthto be negligible, so it is necessary to look for further justification for the use of the product. Such justification must come in the form of evidence for a desirable clinical outcome that cannot be achieved by other means.
A number of reports on the cellular reactions associated with Polyurethane-coatedbreast implants have been cited. In most cases, the implants have been associatedwith a normal inflammatory response, characterised by synovial metaplasia which, ithas been suggested, could have a favourable influence on the rate of capsularcontracture. A slight immunological effect has been seen in animals implanted withpolyurethane, with effects noted on T cell differentiation and natural killer cellactivity. While the significance of these findings is not clear, and some adverseclinical reactions of potential concern have been reported (such as late haematoma or granulomatous reactions), the overall pathological response is acceptable and similar to that seen with other silicone gel breast implants.
Several clinical studies have indicated the superiority of polyurethane-coated breast implants in reducing the occurrence or onset of capsular contracture, compared withsmooth and, in some cases, textured silicone implants. While this association iscommonly acknowledged, more prospective data are needed before the benefit ofpolyurethane-coated implants can be irrefutably demonstrated. In view of theassumption that the coating will degrade completely over time and the limitations ofavailable clinical data, uncertainty exists over the long-term effects of thepolyurethane coating on the rate of capsular contracture. Doubts have also been raised anecdotally by plastic surgeons, as to whether a significant difference in contracture rate exists between polyurethane-coated breast implants and those with a textured silicone surface.
Even if the benefit of polyurethane-coated implants is accepted, EN ISO 14971 andISO/DIS 10993-17 both require that, before the benefit associated with a device canbe considered relevant to a risk assessment, a test of feasibility of risk reduction is required. It is necessary to determine whether the risk can be reduced further atreasonable cost. In this case the risk can be eliminated by omitting the coating orusing another coating material. Alternative products, with textured envelopesconstructed only of silicone elastomer, are available that also claim to reduce theincidence of capsular contracture. No information has been reviewed to address the issue of whether it is possible to manufacture foam with similar physical properties to the one used by Polytech Silimed from a material that is resistant to degradation or that breaks down into non-carcinogenic compounds. From the information availableit is thus not possible to conclude that the use of polyester urethane foam is essential to reduce the incidence of capsular contracture to the desired extent.
Conformity of polyurethane-coated breast implants with ISO/DIS 10993-17 dependson which risk management option is selected for dealing with genotoxic carcinogens.The quantitative risk assessment approach favoured in the USA leads to theconclusion that the risk of cancer arising is around one in a million and thus tolerable. By this method, the product should be considered suitable for use. In the UK, however, public policy rules out this option and suitability for use must be judged on the basis that the risk is outweighed by benefit and reduced to a level that is as low as reasonably practicable. Since some doubt remains over the benefit of the coating and alternatives are available that do not give rise to any carcinogenic hazard, the residual carcinogenic risk, albeit small, cannot be justified.
There is therefore no reason to alter the advice given by MDA in 1994 and 1996 thatpolyurethane-coated breast implants should not be implanted in the UK. Recentevidence is also consistent with the advice that any risk arising from leaving existing polyurethane-coated breast implants in place is likely to be low when balanced against the small risks associated with explantation. Removal of existing polyurethane-coated breast implants is not therefore indicated.
ConclusionsThe following conclusions can be drawn:
polyesterurethane foam can break down in situ to form the genotoxic carcinogen 2, 4-TDA;
recent evidence is consistent with the conclusions reached by the COC in 1991or 1994 that:2,4-toluenediamine (2,4-TDA) should be regarded as a probable genotoxiccarcinogen;there is evidence that small amounts of 2,4-TDA can be released frompolyurethane foam in vitro and in vivo;direct evidence exists that 2,4-TDA is released from polyurethane coatedbreast implants in vivo in women. This leads to the presence of 2,4-TDAin the tissues surrounding the implant. These tissues can metabolise 2,4-TDA and there is therefore a potential carcinogenic risk from this release;information on the breakdown of implanted polyurethane foam in humantissues suggests that breakdown occurs over a period of several years;there were no data on the possible release of harmful substances otherthan 2,4-TDA from implanted polyurethane foam either in vitro or in vivo;
2,4-toluenediamine (2,4-TDA) should be regarded as a probable genotoxiccarcinogen;
there is evidence that small amounts of 2,4-TDA can be released frompolyurethane foam in vitro and in vivo;
direct evidence exists that 2,4-TDA is released from polyurethane coatedbreast implants in vivo in women. This leads to the presence of 2,4-TDAin the tissues surrounding the implant. These tissues can metabolise 2,4-TDA and there is therefore a potential carcinogenic risk from this release;
information on the breakdown of implanted polyurethane foam in humantissues suggests that breakdown occurs over a period of several years;
there were no data on the possible release of harmful substances otherthan 2,4-TDA from implanted polyurethane foam either in vitro or in vivo;
no evidence has come to light to indicate that the 1994 COC conclusions need tobe reassessed;
he risk of cancer arising from the implantation of polyurethane foam-coatedbreast implants has been estimated to be in the region of 1:1,000,000. Whilerisks of this magnitude are often considered to be negligible, this sort of riskassessment model is not considered appropriate for application to genotoxiccarcinogens by the UK Department of Health;
there is no scientific reason to alter the advice published in 1994 and 1996 by the UK Department of Health, based on the COC conclusions, which was:
Polyurethane-coated breast implants pose an unquantifiable (but prabably low) carcinogenic risk. Since suitable alternative products are available, these devices should not be implanted.
The integrity of the silicone shell of the implants is unaffected by the breakdown of the polyurethane foam coating.
The available data show that the amount of 2,4-TDA released decreases with time but suggest that the majority of the exposure occurs during the first 3-4 years post-implantation. There are insufficient data to quantify the rate of degradation with certainty. Since polyurethane-coated breast implants were withdrawn from the UK market in 1991, it is unlikely that explantation of these prostheses will significantly reduce exposure to 2,4-TDA. Although unquantifiable, the risk from leaving polyurethane breast prostheses implanted since 1991 in situ is likely to be low when balanced against the small risks associated with explantation. Any decision to explant must remain a matter of clinical judgement, taking into account thewishes and condition of the patient.
there is insufficient evidence to demonstrate conclusively the long-term benefitof polyurethane-coated implants over other products currently available;
the necessity of using the particular polyesterurethane foam applied to breastimplants to achieve the benefits claimed has not been demonstrated;
In summary, there is insufficient evidence for benefits arising exclusively from theuse of the polyurethane coating to justify exposure to the carcinogenic risk, albeit verysmall, associated with its breakdown in situ. Any use of polyurethane-coated breastimplants in the UK thus remains counter to the risk management policy applied by theDepartment of Health to non-threshold risks.
ReferencesBradley, S.G., Munson, A.E., McCay J.A., Brown, R. D., Musgrove D.L., Wilson s.,Stern M., Luster M.I. and White K.L. Jr, (1994a) Subchronic 10 day immunotoxicityof polydimethylsiloxane (silicone) fluid, gel and elastomer and polyurethane disks in female B6C3F1 mice. Drug Chem Toxicol 17 (3) 175-220.
Bradley, S.G., White K.L. Jr, McCay J.A., Brown, R. D., Musgrove D.L., Wilson s.,Stern M., Luster M.I. and Munson, A.E., (1994b) Immunotoxicity of 180 dayexposure to polydimethylsiloxane (silicone) fluid, gel and elastomer and polyurethane disks in female B6C3F1 mice. Drug Chem Toxicol 17 (3) 221-269.
Brinton, L.A., Malone, K.E., Coates, R.J., Schoenberg, J.B., Swanson, C.A., Daling,J.R. and Stanford, J.L. (1996) Breast enlargement and reduction: Results from a breast cancer case-control study. Plast Reconstr Surg 97 (2), 269-275.
Bryant, H. and Brasher, P. (1995) Breast implants and breast cancer – reanalysis oflinkage study. N Eng J Med 332 (23) 1535-1539.
Bulpitt, K.J., Weiner, S.R. and Paulus, H.E. (1998) Comparison of the systemicmanifestations associated with polyurethane-coated and non-coated silicone breastimplants. Arthritis and Rheumatism, 35 S161.
COM (2001) Statement on Risk Assessment of In-Vivo Mutagens (and GenotoxicCarcinogens). Committee on Mutagenicity Statement – COM/01/S3 – June 2001(www.doh.gov.uk/com.htm)
Council Directive 93/42/EEC of 14 June 1993 concerning medical devices. OfficialJournal of the European Communities; L 169; 1-43 (12 July, 1993).
Delclos, K.B., Blaydes, B., Heflich, R.H. and Smith, B.A. (1996) Assessment of DNAadducts and the frequency of 6-thioguanine resistant T-lymphocytes in F344 rats fed2, 4-toluenediamine or implanted with a toluenediisocyanate-containing polyesterpolyurethane foam. Mutat Res 367 (4) 210-218.
De Lorenzi, E., Massolini, G., Macchia, M. and Caccialanza, G. (1995) HPLCdetermination of urinary 2,4- and 2,6- toluendiamines as potential degradationproducts of polyurethane breast implants. Chromatography 41 (11/12), 661 – 665.
EN 1441; Medical devices – risk analysis; European Committee for Standardization,Brussels (1996).
EN ISO 14971; Medical Devices – Application of risk management to medicaldevices. European Committee for Standardization / International StandardsOrganisation (2000).
Handel, N., Jensen, J.A., Black, Q., Waisman, J.R. and Silverstein, M.J. (1995) Thefate of breast implants: A critical analysis of complications and outcomes. PlastReconstr 96 (7), 1521-1533.
Hester, T, R., Ford, N.F., Gale, P.J., Hammett, J.L., Raymond, R., Turnbull, D.,Frankos, V.H. and Cohen, M.B. (1997) Measurement of 2, 4-Toluenediamine in urineand serum samples from women with Même or Replicon breast implants. PlastReconstr 100 (5) 1291-1298.
ISO/DIS 10993-17; Determination of allowable limits for residues in medical devicesusing health based risk assessment (Draft International Standard); InternationalStandards Organisation, Paris (1999).
Kennephohl, E., Ridley, D., Kerr, A.B. and Daniels, J.M. (2000) Human health riskassessment of diamine isomers from polyurethane foam used in an implantabledevice. Society of Toxicology. Abstract No 506.
LPU Report (1996) Determination of toluene diamine (TDA) under simulatedphysiological conditions with and without the addition of papain.
Lübbers, K. (1997) Polyurethane foam-covered silicone gel-filled breast implants – acritical survey of the literature. For Polytech Silimed Europe Gmbh.
Luke, J.L., Kalasinsky, V.F., Turnicky, R.P., Centeno, J.A., Johnsson, F.B. andMullick, F.G. (1997) Pathological and biophysical findings associated with siliconebreast implants: a study of capsular tissues from 86 cases. Plast Reconstr Surg 100(6), 1558-1565.
Luu, Hoan-My Do, Hutter, Joseph C., Bushar, Harry F. (1998) Physiologically BasedPharmacokinetic Model for 2,4-Toluenediamine Leached from polyurethane Foam-covered Breast Implants. Environmental Health Perspectives, 106, 393 – 400.
Raso, D.S. and Greene, W.B. (1995). Synovial metaplasia of a periprosthetic capsulesurrounding a polyurethane foam breast prosthesis. Ann Plast Surg 35 (2), 201-203.
Santerre, J.P., Labow, R.S., Duguay, D.G., Erfle, D. and Adams, G.A. (1994)Biodegradation evaluation of polyehter and polyester-urethanes with oxidative andhydrolytic enzymes. J Biomed Mater Res 28 (10), 1187-1199.
Sepai, O., Henschler, D., Czech, S., Eckert, P. and Sabbioni, G. (1995) Exposure totoluenediamines from polyurethane-covered breast implants. Toxicology letters 77(1-3), 371-378.
Vázquez, G. (1999) A ten year-experience using polyurethane-covered breastimplants. Aesth. Plast. Surg. 23 189-196
Wang, B.H., Chang, B.W., Sargeant, R. and Manson, P.N. (1998) Late capsularhematoma after breast reconstruction with polyurethane-covered implants. PlastReconstr. Surg. 102 (2) 450-452.
Wang, G.B., Labow, R.S. and Santerre, J.P. (1997) Biodegradation of apoly(ester)urea-urethane by cholesterol esterase: Isolation and identification ofproncipal biodegradation products. J Biomed Mater Res 36 (3), 407-417.
eferences Not Cited But of Potential RelevanceLabow, R.S., Meek, E. and Santerre, J.P. (1999) The biodegradation ofpolyurethane(s) by the esterolytic activity of serine proteases and oxidative enzymesystem. J Biomater Sci Polym Ed 10 (7), 699-713.
Pinchuk, L. (1994) A review of the biostability and carcinogenicity of polyurethanesin medicine and the new generation of “biostable” polyurethanes. J. BiomaterialsScience Polym. Ed. 6 (3) 225-267.
Sampalis, J., Kerrigan, C.L., Hester, R. (1994) Long-term outcome in women withpolyurethane covered breast implants. A proposal submitted to the plastic surgeryeducational foundation. March 1 1994.
Schhnorr et al. 1996
Tsuchiya, T., Takahara, A., Cooper, S.L. and Nakamura, A (1995) Studies on thetumor-promoting activity of polyurethanes: Depletion of inhibitory action ofmetabolic cooperation on the surface of a polyalkyleneurethane but not apolyetherurethane. J of Biomed Mat Res 29, 835-841.
Zhang, y.-Z., Bjursten, L.M., Freij-Larsson, C., Kober, M. and Wesslén, B (1996)Tissue response to commercial silicone and polyurethane elastomers after differentsterilization procedures. Biomaterials 17, 2265-2272.
Is the need for additional breast procedures or revision common after breast enlargement surgery?
The need for additional surgery after primary breast augmentation surgery is high, on order of 1 in 5 to 1 in 4 patients. Multiple factors account for this. Weighing down a breast with a silicone or saline breast implant may increase the chance for needing a breast lift down the road. Capsular contracture, or hardening of the breasts after breast enlargement surgery may require a revision depending on the severity of the problem. The rate of leakage and deflation also goes up with time, increasing the chances for implant removal. Patient request for style/size change, and biopsy are other reasons for additional procedures.
Minimizing Palpability
A breast implant is more likely to be noted on manipulation of the breast when they are too big for the breast and soft tissue present, when they are over, rather than under the muscle, and when they are textured. Ensuring a small enough base width, good soft tissue cover with a submuscular or dual-plane placement, and using smooth breast implants will decrease the risk for this.
What are the various saline implants available from Mentor?
What are Cohesive Gel Breast Implants (Gummy Bear Breast Implants)?
Cohesive gel breast implants were formulated with a thicker form of silicone (viscous silicone polymer as a result of more extensive cross-linking) resulting in three ideal properties; less silicone “run-off,” in the case of rupture, no rippling, and maintenance of more precies form. If you slice into a third generation cohesive gel implant from Mentor and turn the two halves over, the silicone will not leak, drip, or run. You can see how this would be advantageous in case of rupture. It should be mentioned that “cohesiveness” is an attribute that is present in most of the older generation implants, so technically using such implants may be described as using cohesive gel implants. Recently, the latest generation of silicone breast implants was dubbed cohesive in the media. This is true, but misleading, once again, because all silicone implants are cohesive to a certain extent. What most consumers are interested in when they refer to cohesive implants are the so called “gummy bear implants,” or the latest generation of highly cross-linked silicone breast implants. So, back to the two most favorable properties of the so called “gummy bear breast implants.” They are more solid, and they do not ripple. This makes them ideal for thin skinned patients, because soft tissue cover (breast tissue or fat) is not as important in concealing the implant since the formed or cohesive gummy bear breast implant should hold its shape, and not ripple. It is also ideal in breast reconstruction for the same reason, and because large incisions are already present from the mastectomy (breast removal).
Does the FDA publish any information on breast implants?
Breast Implant Questions and Answers (2006)
General Information About Breast Implants
What are breast implants?
What types are approved by FDA?
How are breast implants used?
Are there any age limits with respect to who can get breast implants?
Why is the age minimum different for augmentation for saline-filled and silicone gel-filled breast implants?
Are clinical studies of breast implants still being conducted?
How can I participate in a clinical study?
What are the risks of breast implants?
How long do breast implants last?
What causes breast implants to rupture?
How will I know if my breast implant has ruptured?
If my breast implant ruptures, should I have it removed?
Will the platinum in silicone breast implants harm me?
What are some of the important factors I should consider when deciding whether or not to get breast implants?
What are “gummy bear” gel breast implants? .
How can I report problems with my breast implants?
Where/how can I find the adverse event reports for silicone gel-filled breast implants?
Why do companies file summary reports of adverse events, instead of individual MDR reports for breast implants?
What guidance does FDA provide to the companies that make breast implants?
Details about the approved Silicone Gel-Filled Breast Implants
What was the basis for FDA’s decisions to approve silicone gel-filled breast implants made by Allergan and Mentor? .
Who can get Mentor and Allergan silicone gel-filled breast implants?
What were the most frequent complications seen in the Core Studies for the Allergan and Mentor silicone gel-filled breast implants?
What did the Core Studies show regarding the rate of rupture and the consequences of rupture?
What conditions of approval did FDA place on these implants?
If silicone gel-filled breast implants are safe, why is FDA requiring such large post-approval studies?
What if the post-approval studies uncover unexpected problems?
How will FDA ensure that Allergan and Mentor complete their post-approval studies?
What is the customary cost of breast augmentation repair or breast enlargement revision surgery?
Prices range from as little as $2,500 to as high as $15,000. There are several factors that go into the price of breast enlargement revision surgery.
Geographical area is probably the most likely factor to affect the price of breast enlargement surgery. Prices tend to run lower in regions where the population is less affluent on average, where cosmetic surgery is considered less necessary, but also where competition is more prevalent.
Surgeon availability is another important factor in price setting. A surgeon whose schedule is entirely booked for months on end, will generally “skim the top,” in terms of patients who can afford higher prices. This does not mean there is no one out there who can do as good a job. It means that a particular surgeon knows he or she can get a certain amount of money for his or her breast augmentations.
Another important part of the ultimate cost that is passed on to the patient is whether or not the plastic surgeon owns the facility at which the breast enlargement surgery is carried out. Although part of the facility fee may then go to the owning surgeon, it may be set substantially lower than a facility fee that would be paid to a third party such as a hospital or operating center when carrying out the breast enlargement surgery.
A factor related to the facility fee is whether or not the surgeon prefers to use general anesthesia, or to employ the services of an anesthesiologist, or whether the plastic surgeon favors surgeon administered sedation with regional blocks for carrying out breast enlargement surgery. Obviously, the addition of an anesthesiologist or even a certified nurse anesthetist will add cost to breast augmentation. It is important to realize, however, that paying for an anesthesiologist does add a certain measure of safety, regardless of what anyone may tell you to the contrary.
Can any surgeon use the cohesive “gummy bear” implant?
Any board certified plastic surgeon who has agreed to abide by the patient selection criteria, to provide follow-up data, and to enroll in the ongoing studies may use this implant.
Are “gummy bear” implants the final word in breast augmentation?
This question can be answered by comparing Rembrandt painting with his old brush, and a painter-for-hobby using the latest and greatest in paintbrush technology. It is not so much the brush, as the brusher. I’ll take Rembrandt any day. That being said, the so called “gummy bear” silicone breast implants have many advantages, and a few disadvantages. They are likely the ideal implant for breast reconstruction patients, patients with thin skin or no native breast tissue who desire large breasts, and patients to whom incision size is not of the outmost importance.
Will I be able to breast feed after breast enlargement surgery?
Breast augmentation surgery should not affect breast feeding. Because breasts gain more projection and substance after enlargement, they are typically easier to hold. Because they protrude more, and are easier to hold, breast feeding becomes much easier. Paradoxically breast feeding is actually less difficult for some women, after breast augmentation surgery. Given all of this, however, there are studies showing that women with breast implants report an inability to feed in up to 2/3’s of the implanted population, compared to 7 in 100 for women without breast implants. It is doubtful, however, that matching for size and age such results would ever hold up.
